Fig. 70.1
Purpura (bruises) and petechiae (red and purple dots) on the skin. Bleeding under the skin causes the purple, brown, and red color of the purpura and petechiae (Source: NIH National Heart, Lung and Blood Institute. What Is Thrombotic Thrombocytopenic Purpura? http://www.nhlbi.nih.gov/health/health-topics/topics/ttp)
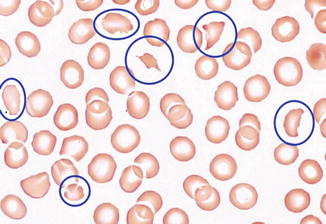
Fig. 70.2
Peripheral blood smear demonstrates schistocytes (circled in blue) (By Central Hematology Laboratory Hemostasis Research Laboratory Bern University Hospital & University of Bern – Own work, CC BY-SA 4.0, https://commons.wikimedia.org/w/index.php?curid=36741808)
Question
What empiric treatment should be instituted urgently for this medical emergency prior to confirmation of the diagnosis?
Answer
Therapeutic plasma exchange
All patients presenting with microangiopathic hemolytic anemia (MAHA) and thrombocytopenia in the absence of an alternative explanation should be recognized rapidly as possible thrombotic thrombocytopenic purpura (TTP). Prompt initiation of therapeutic plasma exchange (TPE) may be life-saving. This patient was admitted to the intensive care unit and a central venous catheter was placed under direct ultrasound guidance without complication following urgent platelet transfusion due to her severe thrombocytopenia. A complete laboratory evaluation was sent including ADAMTS13 level, ADAMTS autoantibody screen, hepatitis panel and HIV testing. Multiple units of fresh frozen plasma (FFP) were administered until TPE could be initiated. TPE was started at 1.5 plasma volumes for the first 3 days. High-dose pulsed intravenous methylprednisolone (1000 mg per day) was administered for the next 3 days. Two units of packed red blood cells and folic acid were also given. Her hemodynamic and cardiopulmonary status was closely monitored. Once her platelet count was greater than in the 50,000/mm3, thromboprophylaxis was started with low molecular weight heparin and aspirin. By day three, her platelet count increased to 150,000/mm3. TPE was then continued at one plasma volume exchanged for two more days and then discontinued. During this time, her ADAMTS13 activity level from admission returned low at 4 %. Hepatitis panel and HIV testing were negative. ADAMTS13 autoantibody was still pending. Her platelet count and LDH continued to improve, and she was downgraded from the intensive care unit by day 10 in stable condition.
Principles of Management
Diagnosis of Thrombotic Thrombocytopenic Purpura
The diagnosis of thrombotic thrombocytopenic purpura (TTP) is primarily a clinical one that is based on the presenting clinical features of MAHA, thrombocytopenia, and organ failure, in the absence of a plausible alternative explanation. A timely diagnosis is essential, because if left unrecognized and untreated, TTP has a mortality rate of 90 % [1, 2]. Clinical presentation of TTP is significantly heterogeneous (Fig. 70.3). The classic pentad of TTP (MAHA, thrombocytopenia, fever, neurologic abnormalities, and acute kidney injury), previously described as occurring commonly, is now quite rare. In one case series of 65 patients with severe ADAMTS13 deficiency, only 5 % were affected by all five of the clinical abnormalities [3]. Prior to advent of early and empiric TPE, patients with TTP would often experience more severe and advanced disease, thus increasing the likelihood of exhibiting the classic pentad prior to death. This likely highlights the greatly improved early recognition of TTP and subsequent initiation of effective therapy which have greatly reduced the risk of developing severe disease with associated multisystem organ failure and mortality. In the same series mentioned earlier, only 37 % had severe neurologic abnormalities and 34 % had no evidence of neurologic dysfunction at all [3]. Acute kidney injury is rare in TTP (seen in only 8 % in that series), as opposed to hemolytic uremic syndrome where it is much more common [3]. Fever was seen in 23 % of the patients with severe ADAMTS13 deficiency and is usually low-grade [3]. A high fever and chills should raise the suspicion for an alternative diagnosis such as sepsis. Since the presence of the classic pentad is now rare and usually present in severe and advanced TTP, clinicians should maintain a high level of suspicion for TTP when only MAHA and thrombocytopenia are found. In these cases, urgent TPE should be undertaken in parallel with an aggressive evaluation for alternative diagnoses. Diseases that may mimic TTP and should be excluded are sepsis, disseminated intravascular coagulation (DIC), autoimmune diseases such as systemic lupus erythematosus (SLE) and scleroderma, vasculitis, malignant hypertension, and malignancy (Table 70.1).
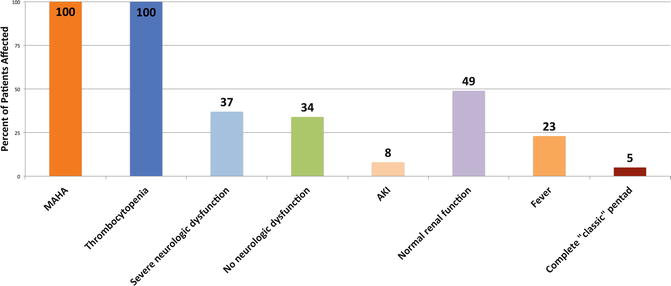
Fig. 70.3
Presenting clinical features in patients with severely reduced ADAMTS13 levels. Data are from day of diagnosis, defined as day of the first plasma exchange treatment. Severe ADAMTS13 activity defined as less than 10 % of normal. Neurologic dysfunction ranged from minor (confusion, headache, etc.) to severe (coma, stroke, seizure, focal signs). Acute kidney injury (AKI) is defined as increased serum creatinine of more than 0.5 mg/dL/day for 2 consecutive days or dialysis together with a serum creatinine of greater than 4.0 mg/dL. Normal renal function is defined as all creatinine values less than 1.5 mg/dL. Complete “classic” pentad of TTP is defined as MAHA, thrombocytopenia, fever, neurologic abnormalities, and renal dysfunction Abbreviations: MAHA microangiopathic hemolytic anemia, CNS central nervous system, AKI acute kidney injury. (Data from: George JN. Diagnosis of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome in adults. UpToDate, 2015; and from George [3])
Table 70.1
Differential diagnosis of thrombocytopenia and microangiopathic hemolytic anemia and their clinical features, ADAMTS13 activity, treatment, and clinical course
Clinical course | 80 % of patients recover; in patients with severe ADAMTS13 deficiency, the relapse rate is 50 %. | Death/ESRD in 12 % of children; 45 % mortality rate in adults; relapse may not occur | Mortality rate is only 7 %; recurrent episodes of bleeding, pancytopenia, infections | Chronic course with a high mortality rate | Favorable with frequent relapses | Relapse may occur, but most subsequent pregnancies are unnaffected | If treated early minimal end-organ damage; recurrence low long term anti-hypertensives | Good outcomes if source control obtained & infection treated early | Depends on staging and type of cancer | Variable, with recurrent thrombosis despite anticoagulation | Mortality is high because of multiple complications | Favorable | High risk of ESRD and death without anticomplement therapy. |
Treatment | TPE, steroids | Supportive care | Steroids or IVIG | Immunosup-pression and TPE | Immunosuppres-sion | Immunosup-pression and TPE | Anti-hypertensives | Antimi-crobials | Surgery, chemo, radiation | Anticoagulation | Immunosuppres-sion | Removal of drug, supportive care | Steroids, TPE, anticomplement therapy |
ADAMTS13 Level | <10 % | <10 % or >25 % | Variable | Variable | Variable | <10 % or >25 % | >10 % | >10 % | >10 % | >10 % | >10 % | >25 % | Variable |
Clinical features | Evidence of ischemic organ injury either in childhood or adulthood | Diarrhea, more common in young children, typically with acute renal failure | Features of TTP with pancytopenia and infectious complications | Severe manifestations of the primary autoimmune disorder, usually including renal failure | Dependent on vessels involved and type of vasculitis | Preeclampsia ore eclampsia, features of HELLP | High blood-pressure, ischemic/neurological symptoms | Features of the primary infection and the organ affected | Features of the primary malignancy | Recurrent miscarriages, with end-organ damnage from arterial/venous clots | Thrombotic microangiopathy usually limited to the kidney | Gradual onset of renal failure over weeks to months | Iniitally presents with acute renal failure in children or adults |
Etiology | TTP: Hereditary or Acquired | HUS | Autoimmune Hemolysis /Evans Syndrome | Autoimmune Disease (lupus nephritis, acute scleroderma) | Vasculitis | Pregnancy-Associated (e.g. HELLP syndrome or eclampsia) | Malignant Hypertension | Infections, either Viral severe bacterial (menor or fungal | Malignancy | Catastrophic Antiphospolipid Syndrome | Hematopoietic Stem Cell Transplant | Medication/drug indueced | Complement-mediated thrombotic microangiopathy |
ADAMTS13 Activity Level and Autoantibody Testing
In 1982, patients with hereditary TTP were observed to have unusually large multimers of von Willebrand factor (ULvWF). This led to the discovery and subsequent characterization of a von Willebrand factor-cleaving protease called ADAMTS13 (or A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13) [3, 4]. In the absence of ADAMTS13, ULvWF multimers released from the vascular endothelium are not cleaved properly resulting in spontaneous platelet-rich microvascular thrombosis in conditions of high shear, such as in the brain, heart, and kidneys with resultant ischemic-induced organ dysfunction. ADAMTS13 deficiency is now well-described to be the causal factor for both hereditary TTP (in association with a confirmed ADAMTS13 mutation) and acquired TTP (in association with an ADAMTS13 autoantibody). Hereditary TTP, also known as Upshaw-Schulman syndrome, is caused by homozygous or compound heterozygous ADAMTS13 mutations, and may be apparent at birth or present later into adulthood when precipitated by a stressor such as pregnancy, infection, or surgery [5]. In acquired TTP, severely reduced ADAMTS13 activity levels of less than 5 % and presence of ADAMTS13 autoantibodies confirm the diagnosis [6, 7] and can help distinguish TTP from HUS with a specificity of 90 % [8, 9]. However, reduced ADAMTS13 activity (greater than 5 %, up to 40 %) may be seen in non-TTP disorders such as uremia, inflammatory states, post-operatively, and during pregnancy [10–12]. In addition to aiding in diagnosis, ADAMTS13 activity levels may help identify patients at increased risk for relapse after treatment. Importantly, definitive therapy with TPE should not be delayed while determining these levels.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


