Chapter 77 Common Endocrinopathies in the Pediatric Intensive Care Unit
Appendix materials for this chapter are available online at http://www.expertconsult.com.
Functional Elements of the Stress Response
The stress response involves an integrated interaction between neurogenic, endocrine, and inflammatory systems, as indicated in Figure 77-1.1–5
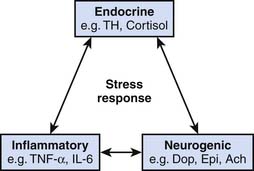
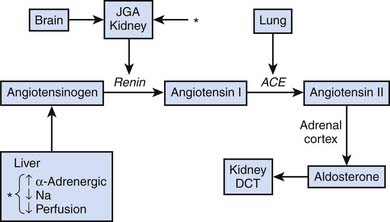
A variety of afferent stimuli that include pain, sight, hearing, smell, pressure changes, chemical alterations, vascular stress receptors, as well as overt inflammation lead to two primary efferent responses: (1) increased sympathetic (and parasympathetic) nervous system activity, and (2) increased release of hypothalamus and pituitary hormones (counter-regulatory response). Catecholamine release inhibits insulin release and action and stimulates synthesis of glucagon, adrenocorticotropic hormone (ACTH), and antidiuretic hormone (ADH). Hypothalamic production of ADH facilitates water retention at the renal distal collecting tubule. Direct neuronal innervation of the kidney and changes in osmolality sensed at the juxtaglomerular apparatus result in activation of the renin-angiotensin-aldosterone axis, ultimately increasing systemic vascular resistance and facilitating sodium retention at the renal distal convoluted tubule, as indicated in Figure 77-2.
Hypothalamic-Pituitary-Adrenal Axis
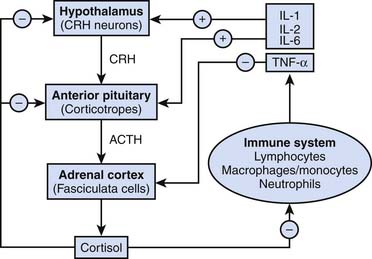
Stressful stimuli lead to activation of the supraoptic and paraventricular nuclei of the hypothalamus, causing the release of ADH and corticotropic-releasing hormone, respectively.3–5 Corticotropic-releasing hormone is transported via hypophysial portal capillaries to the anterior pituitary, facilitating the production of pro-opiomelanocortin, a 239 amino acid peptide that includes primary protein sequences for ACTH, β-lipotropin, β-endorphin, and melanocyte-stimulating hormone. ACTH is transported by the blood to the zona fasciculata of the adrenal gland, where it stimulates the de novo synthesis of cortisol. A schematic overview of the regulation of cortisol synthesis and secretion is provided in Appendix Figure 77-A (see http://www.expertconsult.com).6
Primary activation of the hypothalamic-pituitary-adrenal (HPA) axis in the intensive care unit (ICU) largely reflects the intensity of the systemic inflammatory response syndrome. Interleukin (IL)-6 has been shown to mediate lipopolysaccharide-induced ACTH secretion during infection and inflammation.7
IL-1 and IL-2 also stimulate cortisol production,8,9 whereas tumor necrosis factor-α (TNF-α), macrophage inhibitory protein, and corticostatin (a peptide defensin with anti-ACTH activity) are believed to inhibit it. A general overview of classic endocrinologic pathways affecting cortisol production is shown in Figure 77-3.
Cortisol Biochemistry
The starting point in all steroid hormone synthesis is the conversion of cholesterol into pregnenolone, which represents the rate-limiting step in cortisol production. It requires both mobilization of cholesterol from storage depots and its transfer to cytochrome P-450 located on the inner mitochondrial membrane that catalyzes side chain cleavage. Pregnenolone is then converted to 17-hydroxyprogesterone, and through the enzyme 21-β hydroxylase, it then is converted to 11-deoxycortisol. This process leads to a final key step in cortisol synthesis, the conversion of 11-deoxycortisol to cortisol by the enzyme 11-β hydroxylase, another specific cytochrome P-450 isoenzyme. Generation of a unique hydroxyl group at carbon 11 is essential for both the antiinflammatory and glucose homeostasis functions of cortisol. In addition, cortisol exhibits weak mineralocorticoid activity equivalent to approximately 1% that of aldosterone. A schematic overview of the biosynthesis of adrenal cortical hormones is summarized in Appendix Figure 77-B.10
A diurnal rhythm of cortisol production is noted among healthy individuals, with peak cortisol production typically between the hours of 8 and 9 AM and a nadir of cortisol production typically around midnight mediated by negative feedback mechanisms of the HPA axis. These regulatory mechanisms and diurnal variation are overridden during stress and critical illness because of the continued stimulus of ACTH secretion. Consequently a marked increase in cortisol levels is seen in critically ill patients with normal adrenal function. The physiologic adrenal production of cortisol is approximately 9 mg/m2/day. Stress production of cortisol may reach 200 to 300 mg/day, resulting in a plasma total cortisol level occasionally exceeding 60 μg/dL. The mean and range of plasma cortisol concentrations as a function of age have been reported for normal children and may be found in Appendix Table 77-A.11
Actions of Cortisol
Cortisol is transported from the adrenal gland to various tissues via cortisol-binding globulins, namely transcortin, and albumin. Transcortin has a high affinity for cortisol but a low carrying capacity. The opposite is true for albumin.12
Cortisol diffuses through the plasma membrane by binding to the glucocorticoid receptor that is flanked by heat shock proteins (70, 90, and 56) for transport to the cell nucleus, where cortisol binds to glucocorticoid responsive elements. Once in the nucleus, cortisol modulates the transcription of thousands of genes, perhaps 20% of the entire genome.13,14 Although the majority of corticosteroid action is related to changes in gene transcription, corticosteroids also decrease the stability of messenger ribonucleic acids, ultimately decreasing protein synthesis, including a number of inflammatory proteins.13
Inflammation
Much critical illness is thought to be a product of a disturbance of the balance between systemic inflammatory responses (SIRS) versus compensatory anti-inflammatory responses.15–17 Cortisol concentration is locally increased at sites of inflammation through degradation of transcortin by neutrophil elastase and by local up-regulation of 11 β-hydroxy steroid dehydrogenase, which converts cortisone to cortisol.
Cortisol plays a key antiinflammatory role. It inhibits the internalization of nuclear factor κB (NFκB) into the nucleus by increasing the synthesis of IkB, which traps NFκB in the cytoplasm and thus effectively thwarts the up-regulation and synthesis of a variety of proinflammatory genes and mediators. Similarly, cortisol blocks intranuclear NFκB binding to appropriate deoxyribonucleic acid promoter regions (Figure 77-4). Cortisol exhibits similar activity with nuclear transcription factor activator protein-1.
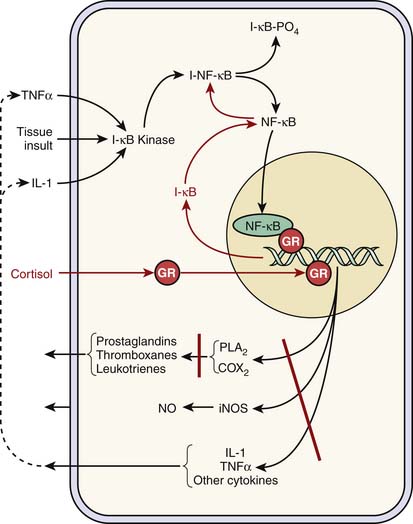
Less well recognized antiinflammatory effects of cortisol include the augmentation of macrophage inhibitory factor, activation of antiinflammatory acute phase proteins including IL-1 receptor antagonist, inhibition of inflammatory cell chemotaxis, down-regulation of adhesion molecules, and the inhibition of neutrophil adhesion and respiratory burst.
Metabolism
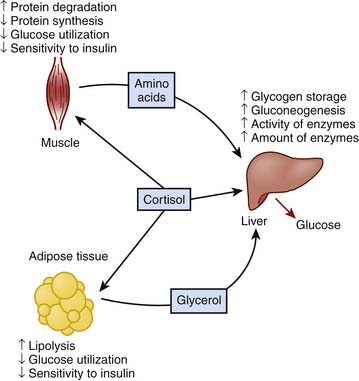
Cortisol promotes energy substrate mobilization through proteolysis, lipolysis, and gluconeogenesis. It exerts permissive actions on glucagon and is antagonistic to insulin. SIRS-activated and cortisol-facilitated lean muscle catabolism provides substrate for hepatic gluconeogenesis and synthesis of acute-phase reactants to cope with the adversity of critical illness but may be inexorably linked to significant corticosteroid adverse effects that may be life-threatening in the ICU. Increased cortisol concentration during stress represents a key mediator in hyperglycemia of critical illness, which will be discussed later. A schematic diagram of the effects of cortisol on metabolism is displayed in Figure 77-5.10
Assessing Adequacy of the Cortisol Stress Response
Historically, adrenal function during critical illness has been ascertained by random baseline plasma cortisol concentrations or by calculating the difference between a corticotropin-stimulated plasma cortisol concentration minus a baseline cortisol concentration. In the latter case a so-called δ value less than 9 μg/dL is considered evidence of inadequate adrenal reserve.18 Depending on the cutoff value chosen (10, 15, 18, 20, or 25 μg/dL), critically ill patients may demonstrate a range of adrenal insufficiency occurrence when random baseline total plasma cortisol concentrations are evaluated.19 Examples of cortisol testing are illustrated in Figure 77-6.
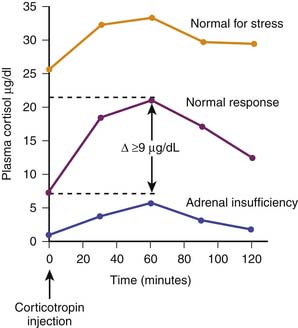
A normal total circulating cortisol concentration is typically in the range of 5 to 10 μg/dL. Patients with true adrenal insufficiency demonstrate concentrations below 5 μg/dL that do not increase with corticotropin or ACTH adrenal stimulation. In the setting of severe stress such as trauma, burns, or sepsis, a normal person typically increases circulating total cortisol twofold to fivefold, generally in the range of 25 to 50 μg/dL. Similarly, a normal person with adequate adrenal reserve will increase circulating total cortisol by at least 9 μg/dL following an exogenous dose of corticotropin. Traditionally, corticotropin stimulation testing has used a corticotropin dose of 250 μg or, alternatively, 145 μg/m2 for children. It has been argued that such a pharmacologic dose of corticotropin may result in a nonsensitive stimulation test, and accordingly, a dose of 1 μg of corticotropin has been suggested.20 However, the latter approach has not been scrutinized as rigorously as the higher dose. For example, more than 50% of a healthy adult cohort demonstrated a δ cortisol concentration less than 9 μg/dL with the low-dose corticotropin stimulation test.21 A detailed evaluation of critically ill adults with sepsis indicated that patients with a high baseline plasma cortisol concentration but a δ cortisol concentration <9 μg/dL exhibited the highest risk for hemodynamic instability and mortality. In fact, a subsequent interventional trial of cortisol was designed based on these findings.22
Although the overnight metyrapone test is not generally applicable to critically ill patients, it has been used as a gold standard in an attempt to identify a population with an apparent inadequate adrenal response to the stress of critical illness. Metyrapone administration will result in an increased ACTH and buildup of the cortisol precursor 11-β deoxycortisol. Among adult patients with severe sepsis, approximately 60% exhibited an abnormal cortisol stress response per the overnight metyrapone test. Best predictors of this situation included a baseline total cortisol concentration of ≤10 μg/dL, a δ cortisol concentration ≤9 μg/dL, or a free cortisol concentration <2 μg/dL.18
Free Cortisol
Although most cortisol is bound to transcortin or serum albumin, the free fraction comprising 10% to 15% of the total is actually responsible for the protean effects of cortisol. Accordingly, it has been suggested that perhaps free cortisol rather than total cortisol might be more reliable in terms of identifying a population that would most benefit from cortisol replacement therapy.23 For most critically ill patients, cortisol-binding globulin is typically decreased and the percentage of cortisol as the free fraction is increased. Moreover, with corticotropin adrenal stimulation, the increase in the free cortisol concentration is greater than the increase in the total cortisol concentration.24,25 Assessing total cortisol concentrations may be especially problematic in critically ill patients with low albumin concentrations.23 Multiple limitations of corticotropin stimulation testing to assess total cortisol concentrations during critical illness have been summarized.26 Free cortisol concentrations may be assessed in urine as well as in saliva,27 but blood contamination of either specimen may limit this approach in the pediatric ICU (PICU). Recently it has been demonstrated that free cortisol concentrations may be determined among critically ill children in real time utilizing temperature-controlled centrifugal ultrafiltration and immunochemiluminescence assay.28 However, what constitutes an adequate free cortisol response among critically ill patients, again with the intent of identifying a population who might benefit from cortisol supplementation, has not been ascertained.
Adrenal Insufficiency in the Intensive Care Unit
Primary Adrenal Insufficiency
Thomas Addison first described primary adrenal insufficiency in his treatise, “On the Constitutional and Local Effects of Disease of the Suprarenal Capsules” in a manuscript published in London in 1855.29 Addison’s disease is more typically encountered in adult patients and is included in a group of disorders termed autoimmune adrenalitis.30,31 Signs and symptoms of Addisonian crisis include intercurrent illness with a history of chronic weight loss and anorexia, dizziness, lethargy, and chronic pigmentation. Symptoms more likely to be associated with admission to the ICU include sudden hypovolemic shock, hyperkalemia, vomiting, diarrhea, abdominal pain, and coma. Other causes of primary adrenal failure include congenital adrenal hyperplasia, with approximately 95% of such cases secondary to 21 hydroxylase (CYP 21) deficiency. In this category, the next most common congenital inborn error of metabolism is 11 hydroxylase deficiency.32 Primary congenital adrenal failure also can occur among infants with adrenoleukodystrophy associated with a metabolic defect in metabolism of very long chain fatty acids. Other causes of congenital adrenal failure include Wolman disease and familial unresponsiveness to ACTH.33
Because of its precarious circulation associated with a subcapsular arteriolar plexus, the adrenal glands are subject to hemorrhage and infarction, particularly in the setting of septic shock—the so-called Waterhouse-Friderichsen syndrome—initially described as adrenal apoplexy.34,35 As indicated in Figure 77-7, a variety of drugs are known to inhibit various metabolic steps along the cortisol synthetic pathway.6
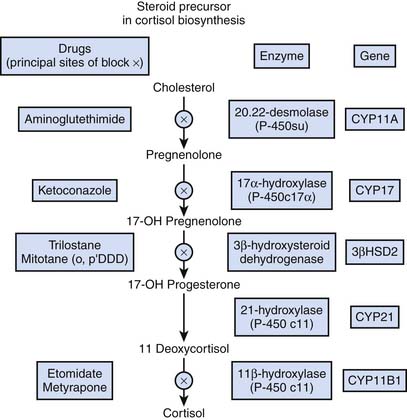
Figure 77–7 Inhibitors of cortisol synthesis.
(Modified from Belchetz P, Hammond P: Diabetes and endocrinology, London, 2003, Mosby Elsevier, p 286.)
Prominent among these inhibitors of cortisol synthesis is etomidate, because it is increasingly being utilized as a sedative for endotracheal intubation to facilitate mechanical ventilation. In one study evaluating patients in an emergency department, more than 50% exhibited a δ cortisol concentration less than 9 μg/dL during a corticotropin stimulation test following etomidate sedation for endotracheal intubation.36 Multiple other investigations report similar findings. In an observational cohort study of 60 children with meningococcal severe sepsis, it was noted that a single bolus of etomidate was associated with impaired adrenal function, specifically 11 β-hydroxylase activity.37 Similarly, an important risk factor for adrenal insufficiency among adult ICU patients requiring mechanical ventilation was use of etomidate for sedation.38 Some experts have advised eliminating the use of etomidate entirely from the ICU39,40 because it may be an independent risk factor for mortality associated with acute adrenal insufficiency.
Secondary Adrenal Insufficiency
Probably the most common cause of secondary adrenal failure seen in the ICU is so-called critical illness–related corticosteroid insufficiency (CIRCI).41 A seemingly inadequate adrenal response relative to the degree of stress characterizes CIRCI, which represents a dynamic, typically reversible situation that is thought to be a result of both decreased cortisol production as well as tissue resistance to cortisol. This resistance to cortisol may result from depletion of corticosteroid-binding globulins, activation of 11β-hydroxyl steroid dehydrogenase and other catabolic reactions, decreased glucocorticoid receptor density and activity, and elevated antiglucocorticoid compounds and receptors.42–45 Currently CIRCI is thought to be best diagnosed by δ cortisol (following corticotropin stimulation) less than 9 μg/dL or a random total cortisol concentration of less than 10 μg/dL.18,41
Multiple pediatric observational studies, most related to sepsis, have correlated random, baseline serum cortisol concentrations with various outcomes.37,46–49 In summary, these studies indicated a loss of ACTH-cortisol circadian rhythm among children with sepsis. As critical illness severity increased (e.g., sepsis → septic shock → death from sepsis), proinflammatory mediators such as IL-6 and TNF-α, as well as ACTH, increased while cortisol concentrations decreased. Both serum cortisol and ACTH concentrations correlated with illness severity per pediatric risk of mortality and organ dysfunction scores, lactate, and C-reactive protein. Three pediatric observational studies also have examined corticotropin stimulation testing among children with severe sepsis.50–52 These studies in general demonstrated that, like adults, an inadequate adrenal reserve is common among children with sepsis. For children with inadequate adrenal reserve, illness severity is increased, as is the requirement for vasoactive-inotropic resuscitation. Such children also more frequently demonstrate vasoactive-inotropic resistance shock and multiple organ dysfunction syndrome. Chronic illness, the degree of organ dysfunction at presentation, and an inadequate adrenal reserve (δ cortisol concentration <9 μg/dL) predicted risk of mortality.
Cortisol Replacement Studies
Adult Investigations
Most clinical investigations related to cortisol replacement therapy in the ICU have focused on patients with septic shock and unstable hemodynamic status despite both fluid and vasoactive-inotropic support. Several investigations examining high doses of methylprednisolone in the 1970s and 1980s found no benefit in terms of reduced mortality but increased risk of significant adverse effects. Subsequently, the notion of relative adrenal insufficiency or CIRCI emerged as a concept. Multiple investigations utilizing hydrocortisone instead of methylprednisolone administered at low stress doses consistently demonstrated hastened resolution from septic shock, and some investigations also reported reduced mortality with this alternative approach. A key hydrocortisone interventional trial was conducted among 19 adult ICUs in France.53 In this investigation, 229 of 300 adult patients with septic shock (74%) demonstrated a δ cortisol concentration less than 9 μg/dL in a standard corticotropin adrenal stimulation test. Among this predefined group treatment, both low-dose hydrocortisone as well as fludrocortisones resulted in faster resolution of septic shock and reduced mortality. However, a follow-up trial, Corticosteroid Therapy of Septic Shock (CORTICUS, ClinicalTrials.gov number NCT00147004), which involved 499 adult patients with severe sepsis, did not confirm these findings. This investigation concluded that hydrocortisone did not improve survival among adults with septic shock but did hasten reversal of shock among patients in whom shock was reversed.54
Pediatric Investigations
Interventional clinical trials examining adjunctive corticosteroids for pediatric sepsis are sparse. Data regarding corticosteroid administration for pediatric Dengue fever are conflicting, and a Cochrane systematic review on this subject concluded no benefit from adjunctive corticosteroids for pediatric Dengue shock, summarizing four trials that enrolled a total of 284 subjects.55 A retrospective descriptive investigation querying the Pediatric Health Information System concluded no benefit of adjunctive corticosteroids for pediatric sepsis, but this investigation was hampered by lack of illness severity data.56 Subsequently a similar retrospective descriptive cohort study has been reported utilizing the database generated during the Researching Severe Sepsis and Organ Dysfunction in Children: A Global Perspective (RESOLVE, F1K-MC-EVBP) trial of activated protein C for pediatric severe sepsis.57 Among a cohort of 477 children with severe sepsis, baseline characteristics did not differ among children who did or did not receive corticosteroids. In particular, their mean pediatric risk of mortality III score and number of dysfunctional organs were similar. Indications for corticosteroid prescription (mostly hydrocortisone) were primarily therapeutic to address shock (89%). The 28-day mortality rate from all causes among children receiving and not receiving corticosteroids was 15% and 19%, respectively (P = .30). Similarly, no difference in mean days of vasoactive-inotropic infusion or mean days of mechanical ventilator support was found.
Current Guidelines for Corticosteroid Prescription in the Pediatric Intensive Care Unit
Septic Shock
The American College of Critical Care Medicine has provided updated recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients.41 Based on expert summary of six randomized controlled trials including a meta-analysis, they concluded that adults with recalcitrant septic shock may benefit from moderate doses of hydrocortisone (200 to 300 mg/day or a similar total daily dose administered by continuous intravenous infusion). Higher doses of corticosteroid equivalents appear to be associated with an increased incidence of adverse events.41 Similarly updated clinical practice guidelines for hemodynamic support of children with septic shock recommend (albeit with no level I evidence) hydrocortisone replacement therapy if a child is at risk for adrenal insufficiency or HPA axis failure, for example, children with purpura fulminans, congenital adrenal hyperplasia, prior recent steroid exposure, and those who remain in shock despite high-dose vasoactive-inotropic and fluid resuscitation.58
Bronchopulmonary Dysplasia
Corticosteroids, typically dexamethasone, also have been utilized to decrease lung inflammation and the risk of subsequent bronchopulmonary dysplasia. In this setting a higher corticosteroid dose was associated with increased risk of neurodevelopmental impairment. Some authors have concluded that corticosteroids should not be administered to neonates with a low risk for bronchopulmonary dysplasia because there appears to be no “safe” window for corticosteroid administration among extremely low birth weight infants.59
Neonatal Hypotension
Hydrocortisone is also commonly used to treat hypotension among neonates. However, this practice may be associated with a higher incidence of intraventricular hemorrhage, periventricular leukomalacia, and death.60 A meta-analysis demonstrated that hydrocortisone increases blood pressure and reduces vasoactive-inotropic requirements among hypotensive preterm neonates. However, the actual long-term clinical benefits of this practice remain unclear.61 Long-term sequelae of hydrocortisone administered to hypotensive neonates are yet to be fully elucidated. Similarly, corticosteroids have been used to improve hemodynamic stability following neonatal/pediatric cardiac surgery. Variable practice precludes generalizations regarding the long-term benefits of this intervention.62–64
Acute Lung Injury/Acute Respiratory Distress Syndrome
Five randomized studies enrolling more than 500 adults have evaluated the role of corticosteroid treatment among patients with acute lung injury and acute respiratory distress syndrome (ARDS). These interventional trials consistently reported that corticosteroid treatment was associated with significant improvement in PaO2/fraction of inspired oxygen (FiO2) ratio, reduction in markers of systemic inflammation, duration of mechanical ventilation, and length of ICU stay. It has been recommended that moderate-dose corticosteroids be considered in the management strategy of adults with early severe ARDS and before day 14 in patients with unresolving ARDS.41 Both methylprednisolone and hydrocortisone also have been used in this regard. Best available evidence suggests optimal initial administration of methylprednisolone as a continuous infusion with a total dose of 1 mg/kg/day.41 Similar data are not currently available regarding children with acute lung injury/ARDS.
Postextubation Stridor
A Cochrane systematic review of 11 trials examining corticosteroids for the prevention and treatment of postextubation stridor in neonates, children, and adults (summarizing 2301 subjects) concluded that the prescription of corticosteroids to prevent or treat stridor after extubation has not been proven to be effective overall for neonates or children. However, this intervention merits further study, particularly for patients at high risk of having the extubation procedure fail. Studies of high-risk adults found multiple doses of corticosteroids initiated 12 to 24 hours before extubation to be helpful.65
Corticosteroid Adverse Effects
Relative to the relative balance of SIRS versus compensatory antiinflammatory response syndrome, excessive steroid administration has been associated with increased risk for hospital-acquired infection among adult patients with septic shock,54 among children receiving hydrocortisone following surgery for congenital heart disease,66 and among adults with trauma treated with corticosteroids.67,68 Subjects enrolled in the CORTICUS trial who received hydrocortisone also exhibited a higher risk of both sepsis and septic shock.54
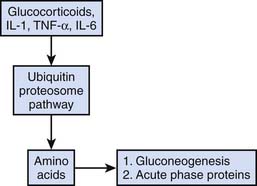
Dissolution of lean body mass mediated by both endogenous and exogenous corticosteroid administration is common in stress states. Although this response may be beneficial in the short term as previously noted, prolonged steroid-mediated muscle catabolism can be associated with prolonged ICU weakness and hyperglycemia. An overview of corticosteroid-mediated proteolysis is displayed in Figure 77-8.69
Prolonged ICU weakness has been associated with prolonged neuromuscular weakness and the need for continued mechanical ventilation even in the setting of resolved lung disease,70–73 whereas hyperglycemia has been associated with a variety of adverse events in the PICU, as detailed in the section of this chapter in which hyperglycemia is discussed. Additional important clinical consequences of protein catabolism that may be exaggerated by exogenous corticosteroid administration include impaired wound healing, hypoalbuminemia, disordered coagulation, and impaired gut function with bacterial translocation.74
In the CORTICUS trial,54 transient hypernatremia also was noted among patients receiving hydrocortisone. Although gastrointestinal hemorrhage represented an important adverse effect in previous clinical trials examining the potential utility of high-dose methylprednisolone as adjunctive therapy in patients with sepsis, this adverse effect has not been problematic in later investigations utilizing low-dose hydrocortisone.
Alterations of Glucose Homeostasis
Normal Glycemic Regulation
Euglycemia occurs when the rate of glucose utilization parallels the rate of glucose production or absorption. Following a glucose load, the body assumes an absorptive state, and its metabolic profile is primarily regulated by insulin. Insulin binds to a tyrosine kinase receptor on cell membranes and triggers a complex series of events that lead to insulin receptor substrate binding, recognition of the activated insulin receptor substrate by intracellular signal transducing proteins, and activation of postreceptor signaling pathways. This action activates phosphatidylinositol-3 kinase and nuclear gene expression modulating proteins. These in turn stimulate glycogen synthesis and inhibit glycogenolysis in liver and muscle, increase glucose uptake by increasing glucose transporter (GLUT)-4 on the cellular membrane, and downregulate the expression of phosphoenolpyruvate carboxykinase (PEPCK) and inhibits fructose 1,6 biphosphatase, the key rate-limiting steps of hepatic gluconeogenesis (Figure 77-9). Insulin also exerts lipogenic and antilipolytic effects.
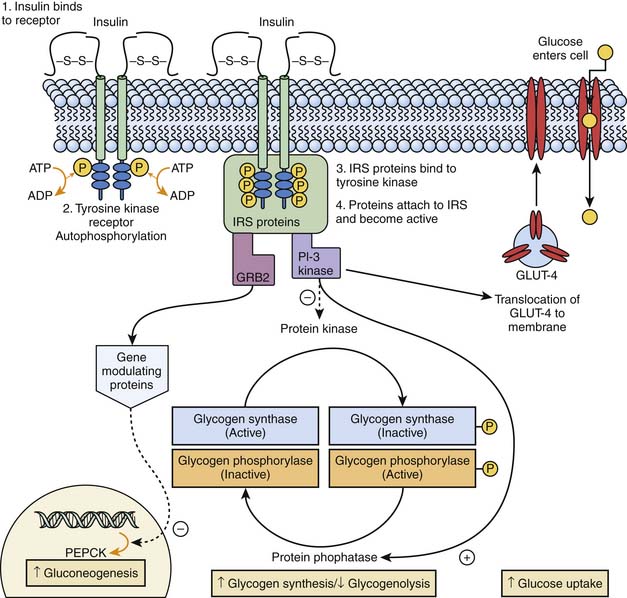
Hormone release in response to serum glucose is regulated by the hypothalamus. It modulates autonomic efferent activity through signals received from peripheral (gustatory, intestinal, and hepatic) and central glucoreceptors. A glycemic rise activates the parasympathetic system and suppresses the sympathetic system, increasing insulin secretion through vagopancreatic stimulation. A drop in blood glucose produces the opposite response, thereby inhibiting insulin secretion and increasing counter-regulatory hormone secretion.75 Euglycemia is maintained not only by a carefully orchestrated and dynamic combination of neuroendocrine mechanisms but also by hepatic autoregulation,76 in which the liver controls hepatic glucose output in response to circulating glucose concentration.77
Hyperglycemia
The American Diabetes Association defines hyperglycemia as a fasting glucose level more than 125 mg/dL. As many as 86% of patients admitted into a PICU were found to exhibit hyperglycemia at some point during their stay.78 Once thought to simply represent an alteration of carbohydrate metabolism in response to severe stress,79 hyperglycemia in critically ill adults and children has more recently received greater attention because of its association with increased morbidity and mortality.80–84
Etiology of Critical Illness Hyperglycemia
During the acute stages of illness there is an increase in glucagon, cortisol, growth hormone (GH), and catecholamine secretion. The resultant increase in gluconeogenesis and glycogenolysis raises serum glucose to levels that would, under physiologic circumstances, be enough to cause a decreased secretion of counter-regulatory hormones and an increase in insulin secretion. In critical illness, however, hyperglycemia is accompanied by persistently elevated levels of proinflammatory cytokines and stress hormones. Normal-to-high insulin levels fail to normalize blood glucose, which reflects insulin resistance. Recent evidence suggests that pediatric patients with cardiovascular and respiratory failure have an absolute rather than a relative insulin deficiency.85 However, it is still unknown whether this deficiency is the result of a primary pancreatic β cell dysfunction in this subset of patients or β cell exhaustion resulting from severe insulin resistance.86
Critically ill patients may demonstrate hepatic and peripheral insulin resistance. In hepatic insulin resistance, high insulin levels fail to inhibit PEPCK expression, and gluconeogenesis continues in the presence of hyperglycemia. The mechanism by which insulin fails to dominantly inhibit PEPCK expression is not known. Although the mechanisms leading to peripheral insulin resistance in critically ill patients are not yet completely elucidated, evidence indicates that the defect lies primarily in the insulin-mediated glycogen synthesis pathway in skeletal muscle87 and not in glucose oxidation.88 Increased levels of counter-regulatory hormones, catecholamines, and inflammatory cytokines, particularly TNF-α, IL-1, and IL-6, have all been linked to the occurrence of insulin resistance.89,90 TNF-α leads to decreased glycogen synthesis and glucose uptake by muscle cells in vitro by impairing insulin postreceptor signaling pathways.89 Additionally, TNF-α induces peripheral resistance to GH,91 leading to decreased insulin-like growth factor-1 levels and reduced inhibition of GH secretion through negative feedback, therefore markedly increasing the release of GH during acute stress. The combination of insulin resistance with high circulating levels of counter-regulatory hormones mediates a catabolic state previously discussed. Concomitant lipolysis causes an increase in free fatty acids (FFAs) that further exacerbates insulin resistance92,93 by altering postreceptor signaling pathways and aggravates the inflammatory response.94





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


