Fig. 36.1
Coagulation pathways . Activation of the extrinsic pathway (through activation of factor VII by tissue factor) and the intrinsic pathway (through contact activation of factor XII) ultimately results in the formation of fibrin (factor I), the building block of the clot. Most steps require the presence of Ca++ and platelet phospholipid. See text for details
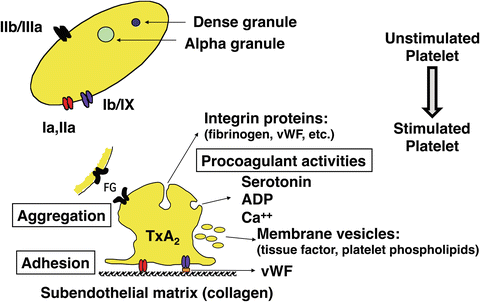
Fig. 36.2
Platelet activation . Platelet activation results in aggregation of platelets and several procoagulant activities. See text for details
The processes described in the previous paragraph are quite complex and are influenced by procoagulant as well as anticoagulant factors while there are also feedback mechanisms with amplifying and inhibiting loops. Although it is clear that clot formation has to be efficient, it is important that these processes are localized and regulated at the same time in order to limit clotting to the site of vessel injury. Indeed, this is essential in order to preserve life because clot formation that goes unchecked would result in massive intravascular clotting. There are several potent mechanisms that normally limit the clot formation to the site of vascular injury, preventing thrombosis in healthy vessels (Fig. 36.3). When thrombin is formed near healthy endothelium, it binds to thrombomodulin expressed on endothelium and thereby activates protein C. With protein S as a cofactor, protein C (both synthesized in the liver and vitamin K dependent) then inactivates factors Va and VIIIa. Protein S has other anticoagulant activities as well [20]. Antithrombin (AT) neutralizes most of the enzymes generated during activation of the clotting cascade, especially thrombin and factors Xa and IXa. The irreversible complex that is formed by the binding of AT and thrombin is thrombin-antithrombin (TAT) and is a sensitive marker of thrombosis. Heparin increases the activity of AT by at least 1000-fold. Heparinoids and heparan (expressed by healthy endothelium) have similar effects as heparin. Prostacyclin and nitric oxide, released by healthy endothelium, inhibit platelet activation, and tissue factor pathway inhibitor (TFPI) , secreted by and expressed by endothelial cells, inhibits factors VIIa and Xa. Finally, plasminogen is activated to plasmin by the release of tissue plasminogen activator (tPA) by healthy endothelium (activated protein C promotes this), and by thrombin, fibrin, and factor XIIa, lysing any fibrin that would have been produced in healthy vessels. Plasmin not only lyses fibrin but also inactivates factors Va, VIIIa, and XIIa. Thus, normal endothelium prevents clot formation by the expression of heparan (activating AT), thrombomodulin (activating protein C), and TFPI, and by the release of prostacyclin, tPA, and TFPI (Fig. 36.3). ADAMTS13 (a disintegrin-like and metalloproteinase with thrombospondin type-1 motifs), synthetized in hepatic stellate cells, cleaves the very large (“hyperactive”) vWF multimers. Deficiency of ADAMTS13 (as a result of severe liver disease or consumption) results in increased platelet activity and various microangiopathies [21]. Disturbances in this complex hemostatic balance can result in hypocoagulation or hypercoagulation.
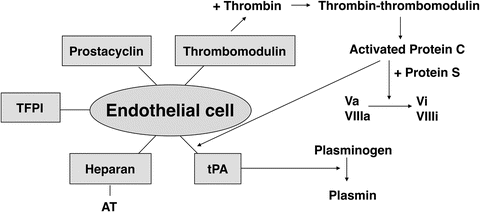
Fig. 36.3
Anticoagulant pathways . The normal endothelial cell plays a crucial role in limiting the clot formation to the site of vessel injury. See text for details. TFPI tissue factor pathway inhibitor; AT antithrombin; tPA tissue plasminogen activator
Fibrinolysis of the formed clot is essential in the eventual restoration of blood flow. This is done through the activation of plasminogen into plasmin, and this process is also controlled by various activators and inhibitors (Fig. 36.4). Activators include tPA, urokinase plasminogen activator, and factor XIIa, and inhibitors include tPA inhibitor (plasminogen activator inhibitor or PAI, released by endothelium), plasmin inhibitor, and thrombin-activatable fibrinolysis inhibitor (TAFI) . Here too, disturbances of this balance may result in hyperfibrinolysis (hemorrhage), or hypofibrinolysis (increased risk of thrombosis).
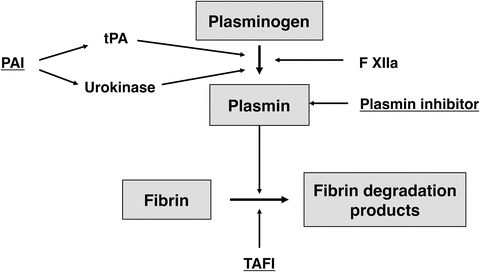
Fig. 36.4
Fibrinolytic system . Activators and inhibitors of the fibrinolytic system. Inhibiting factors are underlined. PAI plasminogen activator inhibitor; tPA tissue plasminogen activator, TAFI thrombin-activatable fibrinolysis inhibitor. See text for details
Coagulation Changes in Chronic Liver Disease (Cirrhosis)
For many years, the impaired synthesis of clotting factors (factors I, II, V, VII, IX, X, XI, XII) by the dysfunctional liver and thrombocytopenia has been considered to result in severe coagulopathy [22]. Vitamin K malabsorption contributes to the coagulopathy through impaired production of factors II, VII, IX, and X. The platelet count is reduced mainly due to splenic sequestration, decreased production (reduced synthesis of thrombopoietin in the liver, hepatitis C infection, folic acid deficiency, and chronic alcohol abuse), and thrombin-mediated platelet consumption [9, 11, 23–25]. There is some evidence of impaired platelet aggregation, reduced adhesiveness, and impaired procoagulant properties of platelets as a result of reduced production of thromboxane A2 and defective signal transduction [26]. Others however have found no evidence of platelet dysfunction [27, 28]. Reduced concentration of factor XIII is seen in a minority of patients with severe liver disease (21 %), and may contribute to increased bleeding [29]. Also, low factor XIII concentration has been associated with increased mortality [29]. The clinical significance of fibrinogen abnormalities (dysfibrinogenemia) is currently unclear [24].
On the other hand, there are changes that enhance thrombus formation: there is a reduction in the concentration of inhibitors of the coagulation system such as protein C, protein S, AT, and tissue factor pathway inhibitor (TFPI) [9, 11, 30, 31], and elevated levels of factor VIII (produced by endothelial cells in kidney, spleen, lungs, and brain) and vWF [32, 33]. Deficiency of ADAMTS13 as a result of severe liver disease results in increased platelet clumping [21]. ADAMTS13 is present in fresh frozen plasma, which is the only available source of ADAMTS13. Here too the increased vWF concentration and reduced ADAMTS13 are rebalanced by a reduction in platelet count (and possibly reduction in platelet function); therefore platelet transfusion should only be done based on bleeding complications, NOT on platelet count [34]. There is evidence for adequate thrombin generation in patients with severe liver disease or undergoing LTx, validating the concept of the rebalancing of the pro- and anticoagulant systems [8, 35].
Changes in the fibrinolytic system mimic the changes in the coagulation system. There is a reduction of both pro- and antifibrinolytic factors: there are decreased levels of plasminogen and alpha2-antiplasmin, but increased levels of tPA and its inhibitor PAI [36]. Although tPA is synthesized by endothelial cells, it is metabolized by the liver, resulting in increased concentrations in liver disease [11, 37]. In most patients this results in a new balance, but in a minority of patients there is increased tPA activity. Some feel that a hyperfibrinolytic state in chronic liver disease, at least in part due to a decrease in thrombin activatable fibrinolysis inhibitor (TAFI, synthetized in the liver) concentration, may contribute to bleeding [38]. Bacterial infection plays a role by stimulating the release of tPA, contributing to a hyperfibrinolytic state [39]. The hyperfibrinolytic state can be documented by the presence of D-dimers [40].
Overall, the simultaneous and opposing changes in both the coagulation and fibrinolytic systems in patients with liver disease are now felt to result in a new balance (Fig. 36.5) [31, 41, 42]. This fragile coagulation balance can be tipped into a state of severe hemorrhage or, less frequently, thrombosis. We have all observed that patients with severe liver disease have episodes of hemorrhage, most frequently intestinal hemorrhage mainly caused by portal hypertension. Also, bleeding may be provoked by stressing factors such as infection or renal failure [42, 43]. In addition, endogenous heparinoids may be released from the endothelium in the presence of bacterial endotoxins, and their clearance may be reduced [44]. Similarly, a state of hyperfibrinolysis can be triggered by inflammatory mediators and endotoxins. However, patients with cirrhosis also have a higher incidence of peripheral thrombosis with thromboembolism and portal vein thrombosis despite abnormal PT or INR [24, 42, 45, 46]. Obviously reduced flow in the portal vein contributes to this complication, but under the right circumstances and with the right triggers, some of these patients become truly hypercoagulable; after all these patients have reduced concentrations of the inhibitors of the coagulation system and increased concentrations of vWF and factor VIII. The release of platelet-derived microparticles by endotoxemia or systemic inflammation may be such a trigger and result in a procoagulant effect by expressing phospholipids and tissue factor; this process is associated with systemic complications and adverse outcome in patients with acute liver failure [47–50]. Intracardiac thrombosis (ICT) has been observed during LTx, reflecting a state of hypercoagulability [17]. It is important to recognize that these patients are not “autoanticoagulated,” as frequently thought in the past [11, 42]. Also, certain types of liver disease are associated with a prothrombotic state. In patients with hepatocellular carcinoma this may be the result of hyperhomocysteinemia [51]. Patients with mild or moderate chronic cholestatic liver disease (primary biliary cirrhosis, primary sclerosing cholangitis) have a mild hypercoagulable state, likely the result of better platelet function (enhanced gpIb/V/IX expression), higher fibrinogen concentrations, and lower degree of hyperfibrinolysis [26, 52].
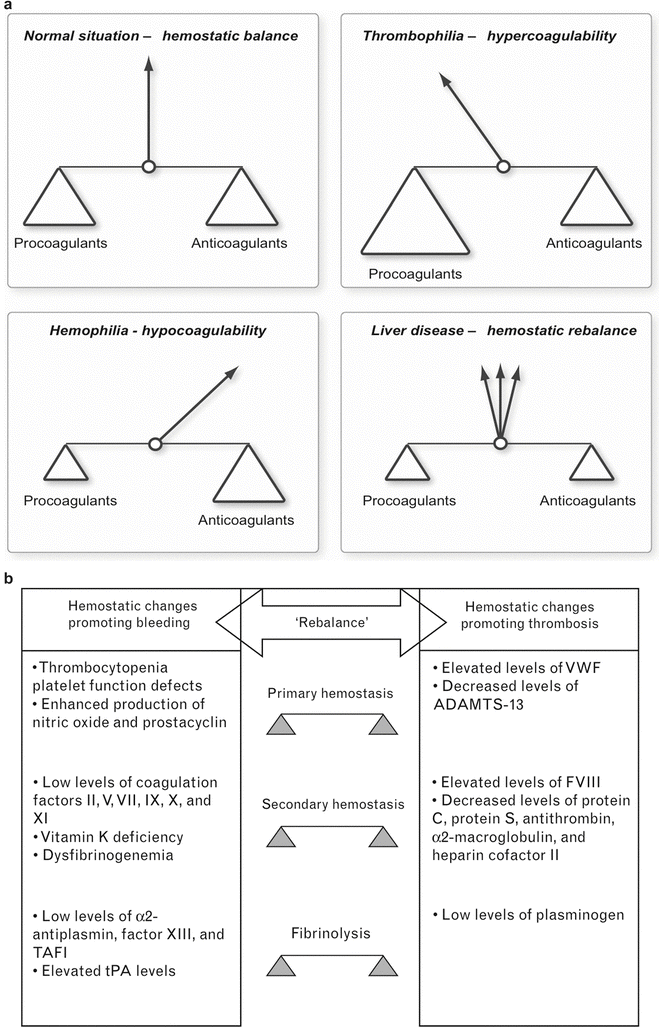
Fig. 36.5
(a) New balance of coagulation in liver disease . Severe liver disease results in a new but more fragile balance of the coagulation system. Relatively small stimuli can result in a state of severe coagulopathy or in hypercoagulability. (b) Changes in pro- and anticoagulant factors in liver disease. Concentrations in both pro- and anticoagulant factors are changed, resulting in a new balance of the hemostatic system
Patients with severe liver disease may have, at the same time, grossly abnormal coagulation tests, signs of accelerated intravascular coagulation, and evidence of fibrinolysis [42]. Although tests frequently suggest the presence of disseminated intravascular coagulation (DIC) , clinically evident DIC is rarely seen, and autopsy results indicate that fibrin depositions are uncommon [53]. It is possible that the high concentrations of D-dimers are the result of the formation of fibrin clot that is more susceptible to fibrinolysis, the increased levels or tPA, or the presence of abnormal fibrinogen; once again, endotoxemia may play a significant role [24, 54, 55].
So, the combination of the reduced concentration of coagulation factors, thrombocytopenia, changes that enhance thrombus formation, and the complex changes in the fibrinolytic system results in a new fragile equilibrium (Fig. 36.5). What are the clinical implications of this new understanding? First, there is a poor relationship between the degree of coagulopathy as determined by the traditional laboratory tests and the occurrence of gastrointestinal bleeding or duration of bleeding after liver biopsy [11, 56]. It is now felt that bleeding from esophageal varices is to a large degree related to a mechanical cause (portal hypertension), while coagulopathy does contribute. Also, blood loss during LTx is poorly related to the degree of abnormality of the traditional coagulation tests [57]. In addition, administration of recombinant factor VIIa shortens the PT results but has minimal clinical benefit during LTx [58]. Thus, coagulation management is better guided by the clinical picture and non-conventional coagulation tests than by the traditional coagulation tests.
Changes in Traditional Coagulation Tests in Severe Liver Disease
The prothrombin time (PT) tests the extrinsic pathway of the coagulation system: it measures the time for the plasma to clot after addition of tissue factor of different origins. PT depends on factors I, II, V, VII, and X, and is a good indicator of severity of liver disease (especially acute liver disease because the extrinsic pathway is dependent on factor VII that has a short half-life). The international normalized ratio (INR) was meant to standardize the effect of the added tissue factor, but there is still inter-laboratory variability. The reduced synthesis of coagulation factors in severe liver disease is reflected in the prolongation of the PT, which is used as an independent marker of severity of liver disease and as a prognostic indicator. The activated partial thromboplastin time (aPTT) tests the contact activation (intrinsic) pathway; it is initiated by activation of the contact factors of plasma. The aPTT is also prolonged in patients with severe liver disease, but usually not as much as the PT. However, both PT and aPTT are poor predictors of bleeding in these patients because of the presence of compensatory mechanisms [41]. Also, these tests are not very sensitive regarding the effects of reduced concentrations of anticoagulants such as AT, protein C, and protein S, and don’t take into account the interactions of the coagulation factors with platelets and the reduced concentration of factor XIII.
Platelet count is reduced in patients with severe liver disease, a result of diminished production in the bone marrow (bone marrow depression and altered thrombopoietin metabolism) and hypersplenism. Platelet function has been assessed by several tests in vitro, but their clinical usefulness has been disappointing [59]. Bleeding time assesses platelet function in vivo, but is highly influenced by factors such as vascular smooth muscle dysfunction in liver disease as well as by variability among those who perform the test. Thus, prolonged bleeding time is observed in up to 40 % of patients with cirrhosis [60], but its clinical relevance remains unclear [9, 28].
Measurements of individual coagulation factors can help in obtaining a more accurate diagnosis, but only the measurement of fibrinogen is of clinical significance to most anesthesiologists. Fibrin degradation products and D-dimers are present in both severe liver disease (as a result of a hyperfibrinolytic state and reduced clearance) and disseminated intravascular coagulation (DIC).
Overall, the conventional coagulation tests (PT, aPTT, INR, platelet count) do not reflect the compensatory mechanisms that play a major role. Therefore, despite abnormal coagulation tests, these patients do not necessarily bleed excessively clinically. Consequently, interventions attempting to normalize these tests may not be required before invasive procedures and during LTx [9].
Monitoring of the Coagulation System During Liver Transplantation
Routine coagulation tests are performed at all transplant centers during LTx, and include at least PT, aPTT, platelet count, and fibrinogen concentration. Many centers use additional tests based on viscoelasticity measurements of clot strength to obtain a better, more complete view of the coagulation system; these tests include thromboelastography (TEG®), thromboelastometry (ROTEM®), and Sonoclot® [61–63]. The main advantage of these viscoelastic tests is that they monitor the coagulation from fibrin formation to clot retraction and fibrinolysis. In addition, these tests are performed on whole blood, allowing the interaction between the plasmatic pathways and platelets, and they can demonstrate the presence of hypercoagulability [64, 65]. TEG® and ROTEM®, and probably also Sonoclot®, provide valuable information on the fibrinolytic process [66].
TEG® and ROTEM® are clotting tests that determine the overall whole-blood coagulability by analyzing the viscoelasticity of the clot during its formation and dissolution [64, 65]. The TEG® analysis includes the effects of most of the factors that affect clot formation, with the endothelium as the only exception. The TEG® tracing displays the torque on a pin that is dropped in a cup containing whole blood that is oscillated 4° 45′ in either direction every 4.5 s at 37 °C (Fig. 36.6). Without clot, the oscillation of the cup has no effect on the torsion wire, and the result is a straight line on the TEG® tracing. When there is clot formation, the maximum rotation of the torsion wire is recorded as the TEG® outline (thromboelastogram). Derived parameters include reaction time (r time; time to 2 mm amplitude, normal range 5–7 min), clotting time (k time; time from 2 to 20 mm amplitude, normal range 1.5–3 min), speed of clot propagation (angle, α, normal range 54–67°), maximum amplitude (MA, normal range 55–73 mm), and clot lysis index (amplitude 60 min after MA is achieved divided by MA, expressed in %). MA reflects the strength of the clot, and a gradual, small reduction in amplitude occurs following the development of the MA and reflects clot retraction, the result of fibrin-platelet interaction in which the platelet’s actin cytoskeleton plays a major role. Clot retraction should be distinguished from hyperfibrinolysis, the result of premature breakdown of the clot by plasmin [67, 68]. The principles of the ROTEM® technique are similar except for a stationary cup and rotating pin, an optical detector system, and the inclusion of an electronic pipette [64, 65]. However, the terminology to describe the tracings is different. Also, because of the different materials that are used, the reference ranges of the TEG® and ROTEM® tracings are not the same [64, 65].
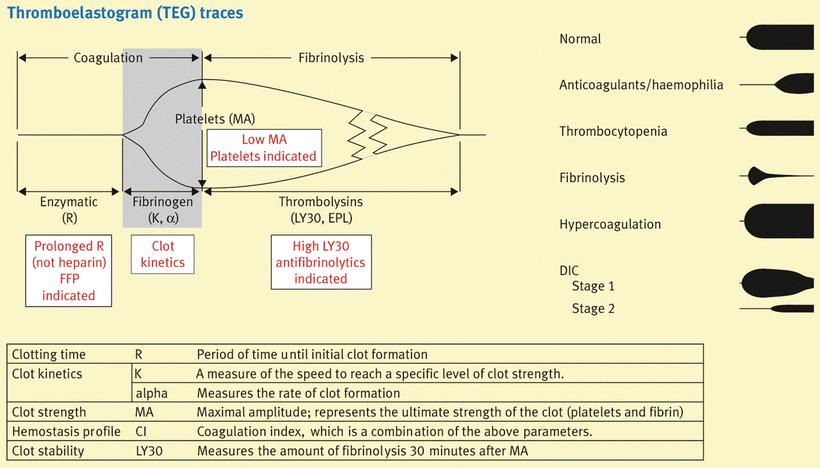
Fig. 36.6
Basics of TEG . Typical thromboelastography tracings for several coagulation abnormalities are presented, and specific therapeutic options for certain TEG abnormalities are suggested
TEG® and ROTEM® allow global assessment of the coagulation system within 20–30 min, including clot initiation (platelet aggregation, platelet-fibrin interaction, fibrin cross-linkage), clot strengthening, clot stability, and eventually clot lysis [69]. Just by looking at the TEG® tracings it can be determined whether the patient has a normal coagulation profile or has hypocoagulability, hypercoagulability, or hyperfibrinolysis. A prolongation of r usually indicates inadequate coagulation factor concentration; a decreased α is usually the result of low fibrinogen and to a lesser degree platelet count or function; and a decreased MA mainly reflects inadequate platelet count or function and to a lesser degree low fibrinogen. Hypercoagulability is reflected by short r time, increased α angle, and increased MA. Hyperfibrinolysis is seen as the rapid narrowing of the TEG tracing (Fig. 36.6). The interpretation of the TEG® can be improved by the addition of specific agents: hyperfibrinolysis in the blood sample can be blocked by adding a small dose of epsilon-aminocaproic acid, while heparin effect can be eliminated by adding heparinase or protamine; the resulting TEG® tracings can then be compared to the native TEG® tracing, allowing detection of hyperfibrinolysis or heparin effect in the patient. The clotting process can be accelerated by adding activators such as celite or tissue factor, and this allows a faster assessment of the coagulation system. However, the interpretation of the test then has to be based on adjusted normal ranges. Celite acts as a contact surface, activating factor XII, and platelets.
Several studies have investigated the relationships between TEG® parameters and traditional coagulation tests; overall the results have been poor [70, 71]. This is to be expected because of the completely different technologies that are used (tests on plasma or isolated platelets vs. whole blood); for example, PT and PTT tests end when fibrin starts to develop. However, when TEG® analysis is performed on platelet-inhibited whole blood or purified fibrinogen solutions, good relationships are found between clot strength (TEG® variable MA) and fibrinogen concentration [72]. Management of the coagulation system has to be based not just on traditional coagulation tests and viscoelastic tests, but should be driven by the clinical need to intervene. Although TEG® and ROTEM® results do not always correlate well with the impression obtained from the surgical field, TEG® and ROTEM® are particularly helpful in determining how to improve clinical coagulopathy.
The viscoelastic tests have several advantages. These are rapid tests, providing us with a good overview of overall coagulability in 20–30 min. They allow the rapid detection of factor deficiency or inadequate platelet count or function, fibrinolysis, and heparin effect. Another advantage is that TEG® includes the effects of factor XIII on clot formation rate and strength (r, α, MA) while the traditional coagulation tests do not [73]. TEG® also allows us to determine the presence of hypercoagulability that cannot be discovered by traditional coagulation tests [70]. Finally, an increase in clot formation rate (as documented by a shortening of the r time on TEG®) has been observed as a result of reduced AT concentration, supporting the concept of the establishment of a new coagulation balance in patients with severe liver disease [74].
Another viscoelastic test is the Sonoclot® . A hollow, open-ended plastic probe is placed in a cuvette with the blood sample. The probe then oscillates vertically in the sample, and the clotting process is reflected in the gradually increasing impedance to movement [64]. Cuvettes with activators or inhibitors are available. Overall there is less experience with the Sonoclot® than with the other viscoelastic tests during LTx [66, 75, 76].
Intraoperative Changes in Coagulation and Their Management
The main causes of bleeding during LTx include portal hypertension, inadequate surgical hemostasis, hypothermia, dilutional and/or consumption coagulopathy, hyperfibrinolysis, effects of synthetic colloids, and release of heparin-like substances and inflammatory mediators from the graft and other tissues. Clamping of portal vein and inferior vena cava (complete or partial) results in an increase in hydrostatic pressure distally in these vessels, contributing to blood loss. The concentration of platelets and most factors affecting coagulation (including fibrinolytic system and inhibitors) will reduce as a result of dilution and/or consumption. It seems logical that severity of liver disease influences transfusion requirements, related to more severe portal hypertension and coagulopathy. However, there is conflicting information on this [77, 78]. A lower transfusion requirement was observed in patients undergoing living donation LTx compared to cadaveric donation LTx, although many factors besides MELD score and degree of coagulopathy could be responsible for this, such as better quality of the graft [79].
Proper coagulation monitoring is essential to manage the coagulation system perioperatively. Certain coagulation problems are associated with the stage of the LTx procedure. For example, surgical bleeding is more common in stages I and II, while hyperfibrinolysis is seen especially towards the end of stage II and early stage III. The most important blood products that are used to correct coagulopathy during LTx include platelets, fresh frozen plasma, and cryoprecipitate. There is limited experience with other blood products such as prothrombin complex and fibrinogen concentrates [80].
Immediate Preoperative Management and Stage I (Dissection)
Because the bleeding risk is not nearly as bad as suggested by the traditional coagulation tests, it is not recommended to attempt to correct abnormal traditional coagulation tests immediately preoperatively or at the beginning of the LTx procedure. To the contrary, the routine administration of fresh frozen plasma, cryoprecipitate, or platelets at that time is expected to increase central venous and portal venous pressure, resulting in increased bleeding caused by increased hydrostatic pressure in the transected vessels [57, 78, 81]. Rather, a low CVP management is chosen by some, necessitating the use of vasoconstrictors to maintain systemic blood pressure [82]. Most anesthesiologists, myself included, however prefer to maintain normovolemia in order to maintain hemodynamic stability, thereby reducing the risk of renal impairment by maintaining its perfusion [83, 84]. Gradual correction of the coagulopathy should be started when intraoperative bleeding occurs and should be guided by coagulation monitoring. If bleeding occurs without evidence of oozing, i.e., surgical bleeding, an argument can be made for the administration of FFP in equivalent amounts to the administration of red blood cells to restore normovolemia and to maintain the fragile, new coagulation balance. Fresh frozen plasma contains not only coagulation factors but also inhibitors of the coagulation system (protein C, protein S, AT, TFPI). Platelet and cryoprecipitate administration are infrequently necessary at this stage, although their administration should be guided by platelet count, TEG/ROTEM, and observation of the surgical field. There is limited experience with prothrombin complex concentrate in liver transplantation [80]. This purified concentrate contains not just factors II, VII, IX, and X, but also protein C, protein S, and AT. Although the administration of prothrombin complex concentrate can normalize PT in patients with liver disease, its efficacy and safety during LTx have not yet been established [80]. Its main potential advantage would be its low volume.
Related posts:
 Preoperative Evaluation and Preparation for Lung Transplantation
Anesthesia and Intraoperative Management of Renal Transplantation
Preoperative Evaluation and Preparation for Lung Transplantation
Anesthesia and Intraoperative Management of Renal Transplantation
 Intensive Care of the Deceased Multiorgan Donor: One Donor, Nine Lives
Intensive Care of the Deceased Multiorgan Donor: One Donor, Nine Lives
 Preoperative Liver Recipient Evaluation and Preparation
Preoperative Liver Recipient Evaluation and Preparation
 Technical Innovation and Visceral Transplantation
Postoperative Care of Heart Transplant Patients
Technical Innovation and Visceral Transplantation
Postoperative Care of Heart Transplant Patients
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


