Patients
Cirrhotics without cardiomyopathy
Cirrhotics with cardiomyopathy
At rest
• Clinical findings:
Hyperdynamic state (few patients)
• Echocardiography:
1. Prolonged isovolumetric relaxation time (mild)
• Electrocardiogram:
QT prolongation (few patients)
• Clinical findings:
Hyperdynamic state (many patients)
• Echocardiography:
1. Prolonged isovolumetric relaxation time
2. Diastolic dysfunction (E/A ratio of ≤1) (systolic dysfunction [rare])
3. Left atrial enlargement
4. Left ventricular Hypertrophy
• Electrocardiogram:
1. QT prolongation (many patients)
2. Bundle branch block and or ST-segment depression (some patients)
During exercise
• Clinical findings:
1. Normal cardiac output increase
2. Reduction in systemic vascular resistance and mean arterial pressure
3. Impairment in aerobic capacity (peak oxygen consumption)
4. Hyperdynamic circulatory state
• Clinical findings:
1. Impaired cardiac output increase
2. Severe reduction in systemic vascular resistance and mean arterial pressure
3. Chronotropic incompetence
4. Abnormal autonomic reflex
5. Severe impairment in aerobic capacity (peak oxygen consumption)
6. Severe hyperdynamic circulatory state
After TIPS
• Clinical findings:
1. Heart failure (rare, and mild)
2. Ascites (rare)
3. Liver and renal failure (rare)
4. Death (extremely rare)
• Clinical findings:
1. Heart failure (more frequent)
2. Ascites (more frequent)
3. Liver and renal failure (more frequent)
4. Death (more frequent)
5. Further prolongation QT interval
After liver transplant
• Clinical findings:
1. Normalization of portal-hepatic hemodynamics
2. Amelioration of cardiac autonomic function after 12 postoperative months
• Electrocardiogram:
Normalizaton of QT prolungation in 50 % of subjects within 12 months
• Clinical findings:
1. Normalization of portal-hepatic hemodynamics
2. Early myocardial depression
3. Early drop in cardiac index and oxygen delivery
4. Normalization of cardiac structure and function by 9–12 postoperative months
• Electrocardiogram:
Normalization of QT prolongation in 50 % of subjects within 12 months
Pathophysiology of Systemic Vascular Resistance and Cardiac Dysfunction
Chronological sequence in which cardiac alterations impact on cardiomyocyte is not fully defined. Multiple electrical abnormalities play a role in the onset of cirrhotic cardiomyopathy (QT interval abnormalities, electrical and mechanical dissociation, chronotropic incompetence) whose development is also linked to autonomic dysfunction (defects in the sympathetic nervous system and vagal impairment) [9, 19].
Prehepatic Sinusoidal Portal Hypertension
Experimental rat model of pre-portal hypertension has elucidated that vasoactive substances (such as ammonia, endotoxin, prostacyclin, serotonin) are released from the intestine when portal pressure is increased, leading to splanchnic vasodilation and further increase in portal pressure [20]. These substances released in the plasma mediate an altered basal contractility and response to beta-adrenoceptor activation in the heart. The presence of portosystemic shunting, further limiting the degradation of these substances by the liver, significantly worsened cardiac performance [21].
Hepatic and Posthepatic Sinusoidal Portal Hypertension
Liver cirrhosis is the result of a series of phenotypic changes in the hepatic stellate cells that make them activated, transformed, and able to produce extracellular matrix components and vasoconstrictive mediators and, finally, to promote liver fibrosis and increased vascular resistance in portal microcirculation, thus causing portal hypertension [22, 23].
In cirrhosis, two fundamental aspects are present: (a) an increased vascular intrahepatic resistance, due to a dramatic decrease of nitric oxide (NO) production in the sinusoidal and postsinusoidal areas [24], and (b) a marked peripheral arterial vasodilation, principally due to an increased amount of vasodilators (NO, prostacyclin, and others) and a decreased amount of vasoconstrictors, in these same vascular districts [25–27]. The marked peripheral arterial vasodilation is initially compensated by an increase in heart rate and cardiac output (hyperdynamic circulation). Indeed, in the early stages of cirrhosis, the sympathetic nervous system and the renin–angiotensin–aldosterone system are greatly activated to compensate arterial vasodilation but, in the late stages, this compensation is lost and the reduced preload (relative hypovolemia) prevails [28, 29].
This is associated with a process of vascular remodeling in the conductive vessels, consisting in a decrease in the thickness and the total area of the vascular wall and in a reduction in the vessel ability to contract, as demonstrated in animal models of cirrhosis [30].
These vascular changes, influencing the afterload, may mask a blunted cardiac response and the diagnosis of cirrhotic cardiomyopathy may be delayed.
The Blunted Cardiac Response
As previously said, in liver cirrhosis, the marked peripheral arterial vasodilatation is compensated through an enhanced activity of the sympathetic nervous system [28, 29]. It is well known that although short-term sympathetic overdrive increases cardiac performance, prolonged stimulation leads to the occurrence of a cardiomyopathy [9]. This causes profound myocardial remodeling and left ventricular hypertrophy characterized by a desensitization of the beta-adrenergic receptor, pro-apoptotic signaling in cardiomyocytes and pro-fibrotic signaling in fibroblasts. The consequence is that in the early phase of cirrhosis, there is an increased cardiac output and a hyperdynamic circulation, whereas in the advanced phases, systolic and diastolic dysfunction with detrimental cardiac consequences ensue [9].
Changes in sympathetic tone and autonomic function affect not only the heart but also the vasculature, the kidneys, and the lungs. Impaired autonomic dysfunction affects about 80 % of cirrhotics impairing renal perfusion and worsening cardiovascular adaptation, especially in those with decompensated liver disease [31, 32]; however, other clinical abnormalities are crucial in determining the blunted cardiac response (Fig. 35.1).
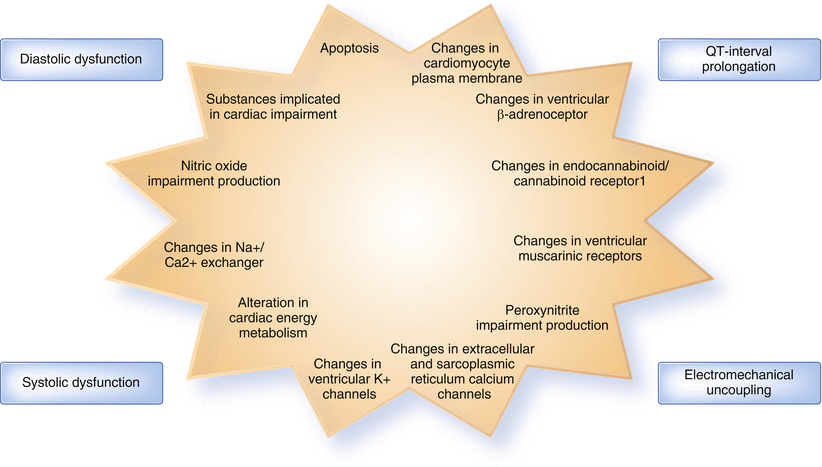
Fig. 35.1
Clinical and pathological basis of cirrhotic cardiomyopathy
QT Interval Prolongation
Prolongation of the QT interval (≥0.44 s), a condition associated with an increased risk for ventricular tachyarrhythmias, is frequently the first abnormality seen in cirrhotics and noncirrhotics with portal hypertension [12]. The length of QT interval also correlates with the degree of liver dysfunction and circulating plasma noradrenaline [12]. K+ channel abnormalities and sympathoadrenergic hyperactivity are both implicated in delaying cardiomyocyte repolarization. Diurnal and nocturnal modifications in autonomic tone circulatory status, and respiratory and oxygen demand, also influence the length of QT interval [33]. After liver transplantation, the QT interval is corrected in nearly 50 % of patients [12].
Electrical and Mechanical Dissociation
Whether QT prolongation mediates the cardiomyopathy or is a marker for the cardiomyopathy is not entirely true. Electromechanical uncoupling has been observed in cirrhotics with QT interval prolongation and it is proposed to be due to defective K+ channel function in ventricular cardiomyocytes [34]. As described above, chronic overstimulation of the beta-adrenergic receptor leads to inotropic and chronotropic incompetence which further promotes electrical/mechanical dissociation [35]. Cardiac diastole is the filling of the heart which occurs as passive flow from the atria to the ventricle during ventricular relaxation: abnormalities in ventricular relaxation, which is an active process, or increased myocardial stiffness, which is a passive process, may lead to diastolic dysfunction. Impaired calcium signaling in cardiomyocyte is seen in cirrhotic [9], and leads to a manifestation of chemical and mechanical molecular changes leading to impaired relaxation. Left ventricular hypertrophy and myocardial fibrosis may lead to increased myocardial stiffness [36, 37].
Cardiac hypertrophy is indeed often seen in cirrhosis and is a result of the hypertrophy of the cardiomyocytes and an increase in the extracellular collagen content and fibrotic reaction [36], and the severity of the hypertrophic response correlates closely with the magnitude of the increased cardiac output [36].
The ratio of early (E wave) to late (A wave) diastolic filling (E/A ratio) and the deceleration time of the E wave are useful echocardiographic Doppler signs measuring the transmitral flow able to evidence diastolic condition after physiological or pharmacological stress test. Unfortunately, the E/A ratio is strongly dependent on preload and in decompensated cirrhotics this may be a disadvantage; furthermore, since diastolic dysfunction may disappear from 6 to 12 months after liver transplantation and cirrhotics with more severe diastolic dysfunction are more likely to have heart failure after liver transplantation [17, 38], it is advisable, in pretransplant phase, to use echocardiographic machines equipped with tissue-Doppler-based algorithm, to obtain more accuracy in detecting diastolic dysfunction, at rest [39].
Although, in several cases of cirrhosis, diastolic dysfunction is of mild degree and does not increase the pulmonary artery systolic pressure to abnormal levels [40], nevertheless it is an independent predictor of mortality and its knowledge helps plan supporting measures to avoid or make more mild the onset of post-liver transplant heart failure [41].
Systolic dysfunction has been associated with alterations in preload, afterload, and diastolic dysfunction. A reduced myocardial reserve, an impaired oxygen extraction (probably due to the local imbalanced NO production and function), and a negative inotropic effect of endocannabinoids participate in causing it [17, 42, 43]. Prolongation in total electromechanical association in systole due to the lengthening of systolic time intervals is the crucial electrical abnormality present in this condition [11]. Cirrhotics may be affected by this condition as demonstrated after pharmacologic or exercise-induced increase in afterload or heart rate in which left ventricular ejection fraction did not change [17, 44].
The echocardiographic evaluation of left ventricular systolic function using new systems as speckle-tracking imaging promises to early identify cirrhotics with functional ventricular impairment [39]. According to some authors, pretransplant risk factors may predict post-liver transplant heart failure. In cirrhotics undergoing liver transplantation, the postoperative onset of systolic heart failure is considered an independent predictor of mortality [41], and identifying these patients before transplantation may optimize perioperative management.
Several pathogenic mechanisms impact on cardiomyocyte membrane and receptor function and give their contribution in causing an impaired myocardial response.
Alteration in cardiac energy metabolism:
Disturbances in substrate utilization seem to have a pivotal role in the pathophysiology of cardiac function. It seems that metabolic changes may precede structural and functional changes in the heart [45]. Dysregulation in cardiac energetics is viewed as one of the potential mechanisms underlying cirrhotic cardiomyopathy [46].
Substances implicated in cardiac impairment:
Carbon monoxide, hemoxygenase, NO, and endogenous cannabinoid bile acids in concert with other circulating molecules (cytokines, endotoxin, chemokines, lipids, for example) have a strong role in impairing cirrhotic heart, thus contributing to induce cardiac changes viewed in cirrhotic cardiomyopathy [47].
Changes in cardiomyocyte plasma membrane:
Changes in the membrane result in the increase in cholesterol content and in the cholesterol-to-phospholipid molar ratio; the principal effect, observed in rat models of cirrhosis but not in portal hypertensive rats, is the reduction of membrane fluidity, the increase of its stiffness, and the lower cAMP production in response to adrenergic stimulation [48, 49] (Fig. 35.2a).
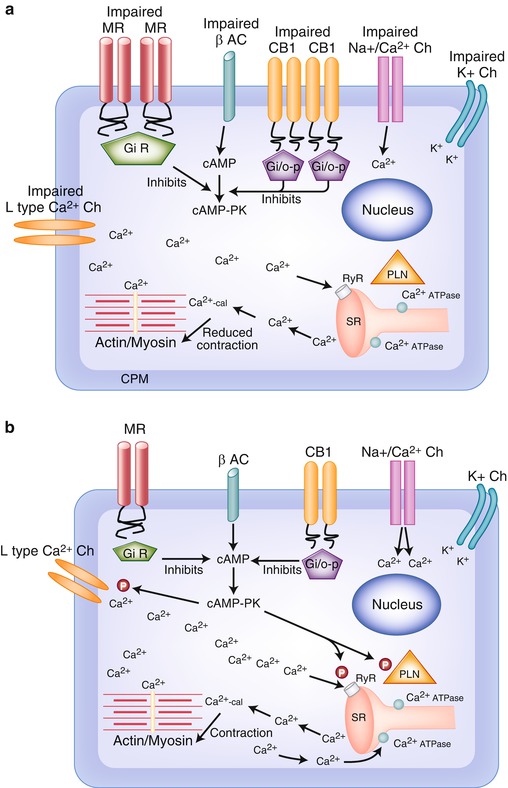
Fig. 35.2
(a) Impaired function of cardiomyocyte in cirrhotic cardiomyopathy . The entry of Ca2+ through L-type Ca2+ channels, in the cardiomyocyte, is imbalanced. The trigger of Ca2 to release Ca2+ from intracellular stores by ryanodine receptor (RyR) in the sarcoplasmic reticulum is low. The contraction is reduced. B-adrenergic receptor is imbalanced and does not contribute to stimulate Ca2+ influx. Reduced Ca2+ influx through L-type Ca2+ channels causes decreased Ca2+ entry and SR load resulting in smaller Ca2+ transients that causes a decrease in contractile force. Abbreviations: β AC β-adrenoceptor, Ca 2+ -cal Ca2+-calmodulin, cAMP adenylyl cyclase, cAMP-PK cAMP-dependent protein kinase, CB1 cannabinoid receptor 1, CPM cardiomyocyte plasma membrane, Gi/o-p Gi/o proteins, K + potassium, K + Ch potassium channels, L-type Ca 2+ Ch L-type Ca2+ channels, MR muscarinic receptor, Na + /Ca 2+ Ch Na+/Ca2+ channels, N nucleus, P phosphorylation, PLN phospholamban, RyR ryanodine receptor, SR sarcoplasmic reticulum. (b) Normal function of cardiomyocyte. Ca2+ influx enters through L-type Ca2+ channels in the cardiomyocyte. Ca2 triggers a further release of Ca2+ from intracellular stores by ryanodine receptor (RyR) in the sarcoplasmic reticulum. Ca2+ influx and released Ca2+ directly favor the contraction. Contraction is terminated by the rapid uptake into the SR by SR Ca2+ ATPases. β-Adrenergic receptor stimulation increases inotropy by phosphorylation (P) of PLN and L-type Ca2+ channels through cAMP-PK-stimulating Ca2+ influx and Ca2+-pump activity. This increases the load of Ca2+ in the SR stores and leads to enhanced Ca2+ transients upon depolarization. Abbreviations: β AC β-adrenoceptor, Ca 2+ -cal Ca2+-calmodulin, cAMP adenylyl cyclase, cAMP-PK cAMP-dependent protein kinase, CB1 cannabinoid receptor 1, CPM cardiomyocyte plasma membrane, Gi/o-p Gi/o proteins, K + potassium, K + Ch potassium channels, L-type Ca 2+ Ch L-type Ca2+ channels, MR muscarinic receptor, Na + /Ca 2+ Ch Na+/Ca2+ channels, N nucleus, P phosphorylation, PLN phospholamban, RyR ryanodine receptor, SR sarcoplasmic reticulum
Changes in ventricular β-adrenoceptor:
Rat models of cirrhotic cardiomyopathy have demonstrated to have a blunted cardiac response due to a decreased membrane fluidity and to an attenuation of the portion of the beta-adrenergic receptor signaling pathway upstream of adenylyl cyclase [50, 51] (Fig. 35.2a).
Changes in ventricular muscarinic receptors:
In rat models of cirrhosis, blunted muscarinic (M2) responsiveness and defective signal transduction to cAMP are reported; interestingly, this was not caused by receptor down-regulation but by changes in postreceptor system of cardiomyocyte [52].
Changes in ventricular K + channels:
Activation of ventricular K+ channels promotes hyperpolarization and relaxation, whereas inhibition causes depolarization and contraction. A decreased K+ current density for the three types of K+ channels present in ventricular myocytes was found in a rat model of cirrhosis [53]. These rats exhibited a longer duration of baseline action potential as compared with ventricular myocytes of sham-operated rats [53]; the QT interval prolongation present in cirrhotics was hypothesized to be a direct consequence of these changes [9]. In another animal study, it was ascertained that the altered inotropic effect was due to changes in K+ channels able to modify resting membrane potential and action potential waveform and thus the intracellular Ca2+ concentration, the key driver of the myocardial contractility [54].
Changes in membrane and sarcoplasmic reticulum calcium channels:
Cardiac myocytes of a rat model of cirrhosis were demonstrated to have a decreased cardiac contractility, due to a decrease in initial plasmalemma Ca2+ entry (through L-type Ca2+) as well as a decreased Ca2+-stimulated Ca2+ release, whereas intracellular systems were showed to be intact [55] (Fig. 35.2a).
Changes in Na + /Ca 2+ exchanger:
Na+/Ca2+ is responsible for maintenance of a steady-state intracellular free Ca2+ concentration and its impairment may thus contribute to the onset of cirrhotic cardiomyopathy [56, 57].
Changes in endocannabinoid/cannabinoid receptor1:
Studies in vitro and on rat models of cirrhosis indicated that the increased activity of cannabinoid receptor1 may be responsible for myocardial contractility dysfunction [42, 58].
NO impairment production:
An overproduction of NO is cardiodepressant, but inducing a splanchnic arterial vasodilation and a hyperdynamic circulation in cirrhosis masks the presence of blunted cardiac function [59] (Fig. 35.2a).
Peroxynitrite impairment production:
An overproduction of peroxynitrite depresses cardiac function through nitration (or S-nitrosation) of cardiac contractile proteins, such as actin [60].
Apoptosis:
Cardiomyocyte apoptosis may play a pivotal role in myocardial remodeling in heart failure [61]. Several lines of evidence indicate that also a mild increase of the apoptotic rate may induce cardiac dysfunction; changes in mRNA and protein expression levels of tubulin and collagen characterize this phase [62]. Interestingly, apoptosis (cardiac damage and fibrosis) has been appreciated in experimental animal models in preclinical cardiac failure [62].
Treatment and Prognosis
Cirrhosis is a life-threatening condition with an overall unfavorable prognosis if liver transplant is not safely performed. Impaired cardiac reserve is an additional condition that may worsen the prognosis especially in the setting of acute decompensated cirrhosis. Increased cardiac output, indeed, serves as an important compensatory mechanism in cirrhosis, and loss of the compensation may be critical in determining patient discharge in conditions of further reduction in systemic vascular resistance (such as sepsis) or reduced intravascular volume (such as hemorrhage) [63]. The inability to increase cardiac output, favoring a decrease in renal perfusion, likely contributes to the pathogenesis of hepatorenal syndrome driving to unfavorable outcomes [11, 64]. The ensuing sympathetic activation increases cardiac contractility but also stimulates renal sodium and water retention through the activation of the renin–angiotensin–aldosterone system. Overactivation of the sympathetic tone and the renin–angiotensin–aldosterone systems contributes to worsening cardiac and renal remodeling and dysfunction, thus, negatively, affecting the prognosis [65]. Liver transplant, completely changing the natural history of the disease, may be the cure for cirrhosis and the associated cardiomyopathy [9].
There are no specific treatments for cirrhotic cardiomyopathy. In the absence of dedicated studies, general knowledge and considerations used for heart failure have been considered, with some exceptions. Angiotensin-converting enzyme inhibitors (and vasodilators in general) are a mainstay in the treatment of systolic heart failure, but they likely have little or no role in the cirrhotic cardiomyopathy, a condition with which there is severe systemic vasodilation. Beta-adrenergic blockers have also shown to reduce mortality in patients with systolic heart failure. Data on beta-adrenergic blockers in patients with heart failure and preserved ejection fraction are not available; yet such drugs are often used. There is a clear rationale to use beta-adrenergic blockers in patients with cirrhotic cardiomyopathy since their sympathetic overdrive is the key feature of the disease process; the use of these drugs is sometimes hindered by hypotension, though needed for the prevention of recurrent variceal bleed. A selective beta-blocker (beta1 and beta2) without alpha-blocking activity may be preferred to avoid further vasodilation, although some experimental studies show favorable data with carvedilol, a nonselective alpha-beta1/2 blocker. Appropriately sized controlled trials with beta-blockers in this disease are however lacking. Diuretics including loop-diuretics and aldosterone antagonists are often used to treat hypervolemia in cirrhotics with and without cardiomyopathy, but whether such treatment affects the outcome is unknown [9].
After liver transplantation, there is both a rapid hemodynamic change and an increased filling pressure that may worsen a pre-existing congestive heart failure. Some studies demonstrated cardiovascular complications in almost 25 % of patients undergoing liver transplant and a higher risk for postoperative pulmonary edema in patients with an abnormal heart function [66, 67]. Since conflicting results exist on the correlation between cirrhotic cardiomyopathy and severity of liver disease [63, 68, 69], all cirrhotics, independently of their Child-Pugh-Turcotte or model for end-stage liver disease (MELD) classifications, should be screened to evaluate the presence of cardiac abnormalities in pretransplant phase. An interesting study performed on 64 cirrhotics screened before liver transplant demonstrated that 23 % (15 patients) was affected by cirrhotic cardiomyopathy [68].
The fact that an amelioration of cardiac function has been observed after liver transplant corroborates the concept that the cardiomyopathy is truly cirrhotic in origin [38] and that pretransplant evaluation of cardiac function is necessary to better plan the management of the patients and their long-term outcome.
Pretransplant Investigation of Cirrhotic Cardiomyopathy
Comorbid conditions of the recipient influence the outcome of liver transplant [70]. Increasing rates of cardiovascular complications after liver transplantation have been observed. They affect over 70 % of liver allograft recipients but cardiovascular mortality is reported to be less than 7–15 % [71]. Child-Pugh or MELD classifications are able to evaluate the severity of liver disease but fail to predict the post-liver transplant outcome [72]. Therefore, cardiac evaluation may be an additional screening tool to predict cardiovascular complications in the post-transplant period. The New York Heart Association (NYHA) and the Framingham score are useful tools to identify cirrhotics with heart failure but do not provide specific recommendations for the pretransplant assessment of liver transplant candidates.
It is known that the presence of an hyperdynamic circulation in cirrhosis is directly related to the severity of hepatic disease [73]. In conditions of marked vasodilation, if a high cardiac output cannot be maintained by a good cardiac function, the prognosis is grave [73]. Monitoring cardiac function may therefore be useful, and several noninvasive assessment tools exist. Electrocardiography and echocardiography are performed in most transplant liver centers to identify individual characteristics and cardiac compliance to liver transplant. The presence of cardiovascular complication after liver transplantation raised also the question of the underlying reasons for heart failure and electrical abnormalities. Cirrhotic cardiomyopathy with overt left ventricular failure has emerged as an important cause of perioperative morbidity and mortality for liver transplant recipients [16].
Cirrhotic cardiomyopathy (variably defined) is present in up to 50 % of the patients undergoing liver transplant. The cardiomyopathy is primarily characterized by electrophysiological abnormalities, a normal cardiac function at rest, and a blunted cardiac response to stimuli and systolic and diastolic dysfunction. The QT interval prolongation is one of the major electrophysiological abnormalities and is related to the severity of the liver disease and the degree of portal hypertension; it may be associated with an increased risk of ventricular arrhythmias but its more severe complication (sudden death) is not a frequent event in cirrhotics [74]. Prolonged QT interval should be evaluated in order to correct the reversible causes, such as electrolyte disturbance or the use of QT interval-prolonging drugs [75].
In spite of different opinions on the relation of cirrhotic cardiomyopathy with the severity of liver disease [63, 68, 69], a consensus exists about its linkage with the hemodynamic changes associated with portal hypertension; the subsequent hyperdynamic circulation impacts on cardiac load and makes more difficult an accurate evaluation of systolic and diastolic function through the conventional echocardiography [74]. Therefore, since systolic and diastolic functions deeply depend on changes in cardiac load, interpretation of conventional echocardiographic images can be challenging. However, a preoperative conventional echocardiography should be performed routinely to evaluate left and right ventricles and valvular function. The American Association for the Study of Liver Diseases recommends transthoracic echocardiography with Doppler for all liver transplant candidates.
A prevalence of left ventricular hypertrophy was appreciated in 30 % of subjects before liver transplant [76]. An interesting echocardiographic study before and within 6 months after liver transplant showed that the presence of diastolic dysfunction (defined as E/A ratio of ≤1) in pre-liver transplant recipients was associated with an excess risk of heart failure and was able to predict post-liver transplant survival [77]. In pretransplant phase, the presence of heart failure should be monitored and optimization of therapy, using beta-blockers and diuretics (including aldosterone antagonists), should be made. Angiotensin-converting enzyme inhibitors should be used sparingly before transplantation and added gradually after transplantation. Successful liver transplant in patients with ejection fraction as low as 10 % has been obtained after aggressive medical management [78].
Tissue Doppler imaging (TDI) , directly measuring the velocity of myocardial displacement, overcomes the impaired preload and afterload present in cirrhotics with hyperdynamic circulation and is considered the most sensitive and reproducible echocardiographic technique for assessing left ventricular filling dynamics; the tissue velocity, measured at the basal part of the lateral and septal left ventricular wall during early filling (E′), is primarily determined by the relaxation of the left ventricle. TDI velocities have significant correlation with invasive indices of the left ventricular relaxation and the E/E′ index is considered the most important parameter of the left ventricular diastolic function [79, 80]. Exercise tests, reproducing a condition of stress as that induced by liver transplantation, may disclose abnormal cardiac reserve or reveal comorbid conditions.
Related posts:
 Preoperative Evaluation and Preparation for Lung Transplantation
Anesthesia and Intraoperative Management of Renal Transplantation
Preoperative Evaluation and Preparation for Lung Transplantation
Anesthesia and Intraoperative Management of Renal Transplantation
 Intensive Care of the Deceased Multiorgan Donor: One Donor, Nine Lives
Intensive Care of the Deceased Multiorgan Donor: One Donor, Nine Lives
 Acute Liver Failure: Perioperative Management
Postoperative Care of Heart Transplant Patients
Anesthesia for Composite Tissue Allografts
Acute Liver Failure: Perioperative Management
Postoperative Care of Heart Transplant Patients
Anesthesia for Composite Tissue Allografts
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


