Chapter 74 Cellular Respiration
In 1920, Haldane was credited with the observation that hypoxemia not only stops the [respiration] machine, but wrecks the [respiration] machinery as well. Indeed, the priorities of pediatric advanced life support are to avoid shock and respiratory failure, and the focus of cardiopulmonary resuscitation is restoration of airway, breathing, and circulation, all targeted to maintain or reestablish oxygen delivery to tissues and cells. During critical illness, marginal oxygen delivery and/or oxygen consumption frequently manifest as the rate-limiting step for efficient energy production in the form of adenosine triphosphate (ATP). Inadequate ATP impairs the translation of cellular structure to cellular function, a defining characteristic of life.1 A working knowledge of cellular respiration basically summarizes the key tenets of critical care medicine. This chapter reviews the metabolism of respiration, summarizes various modalities for monitoring respiration, and provides clinical correlation of alterations in respiration that are addressed by intensive care.
Metabolism of Respiration
Oxygen Chemistry
It is appropriate to initiate a discussion of respiration by describing the relevant characteristics of the rate-limiting substrate, namely molecular oxygen, that serves as the terminal electron acceptor in cellular respiration.1 Because of the paramount role of oxygen in facilitating efficient production of ATP, oxygen has played a central role in terms of evolution of complex multicellular life.2 In the biosphere, the concentration of oxygen is carefully regulated by the processes of photosynthesis and respiration, the former producing—and the latter consuming—atmospheric dioxygen. Premolecular oxygen appeared on Earth’s surface approximately 2 billion years ago and now represents the most abundant element on Earth’s crust. At its baseline triplet state, oxygen is a diradical with two unpaired electrons with parallel spin occupying the outer orbital. In its role as the ultimate electron accepter in cellular respiration, molecular oxygen undergoes a four-electron reduction to water. With the benefit of multiple redox electron exchanges, this process permits controlled release of energy from carbohydrate, fat, and protein energy substrates. Successive one-electron additions to molecular oxygen result in the production of superoxide anion, hydrogen peroxide, hydroxyl radical, and water, respectively (Figure 74-1). These partially reduced oxygen compounds are referred to as reactive oxygen species and identify the so-called antagonistic pleiotropy characteristic of oxygen.3
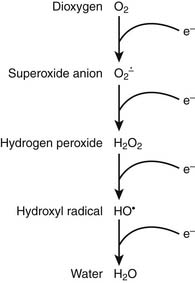
Oxygen toxicity was first predicted by both Priestly and Lavoisier in 1729. It has been estimated that approximately 2 billion molecules of superoxide anion and hydrogen peroxide are produced per cell per day. An antioxidant repertoire that includes superoxide dismutase, catalase, glutathione peroxidase, uric acid, vitamin C, and vitamin E counteracts this oxidant stress to minimize a tendency toward “rust and rancidity.” However, oxidant stress plays an important role in generalized aging as well as inflammation and ischemia-reperfusion pathophysiology. As Fridovich8 has noted, “the aerobic lifestyle offers great advantages but is fraught with danger.”
Nitrogen chemistry is also involved in the generation of reactive molecular species. Nitric oxide (NO) is known to be generated from at least four NO synthase isoenzymes, including constitutive, inducible, neuronal, and mitochondrial forms. Figure 74-2 depicts the reaction catalyzed by nitric oxide synthase.9
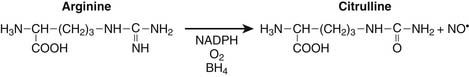
Production of NO is particularly prominent during periods of inflammation. At least in septic patients, intensity of inducible NO production as measured by blood or urine end catabolic products nitrate and nitrite, is directly associated with various measures of illness severity reflecting multiple organ dysfunction syndrome and death. Particularly in the setting of tissue hypoxia, NO is believed to play a role in regulation of mitochondrial respiration through nitrosyl complexes with various iron-sulfur center enzymes as well as direct competition with oxygen for binding at the heme active site of cytochrome oxidase.10,11 Competitive inhibition of NO with oxygen at cytochrome oxidase results in decreased oxygen consumption, decreased binding affinity for oxygen, and reduced ATP production.
Under specific conditions superoxide anion and NO can condense to form the powerful oxidant peroxynitrite with diffusion-limited kinetics: 7 × 109 M−1 × sec−1.12 Behavior of NO, superoxide anion, and peroxynitrite in clinical biochemistry has been characterized as “the good, the bad, and the ugly.”13 For example, nitrotyrosine and dityrosine represent tissue markers of peroxynitrite histopathology.14 Peroxynitrite has been demonstrated to be involved in lipid peroxidation of cell and organelle membranes, damage to various elements of the mitochondrial electron transport chain and ATP synthase, inhibition of glyceraldehyde 3-phosphate dehydrogenase, injury of the sodium-potassium ATPase pump, disruption of plasmalemma sodium channels, production of DNA strand breaks, and activation of the poly-adenosine ribosyl phosphate system.15 Again in reference to cellular respiration, peroxynitrite can mediate irreversible inhibition of the Krebs cycle enzyme aconitase; mitochondrial complexes 1, 2 and 3; cytochrome oxidase; ATP synthase; and creatine kinase as well as increase wasteful proton leakage. Reduced suflhydryls represent another important target of peroxynitrite oxidation, resulting in alteration of protein structure and function.16 Peroxynitrite is known to directly alter pulmonary surfactant and injure human myocardial protein.14,17
Reactive oxygen and nitrogen species are also known to affect intracellular signaling through a number of redox-sensitive transcription factors, notably nuclear factor-κB (NFκB) and activator protein-1 (AP-1).18 For example, as an aspect of the pathophysiology of gram-negative sepsis, interaction of endotoxin, toll-like receptor-4, and NADPH oxidase result in an increased flux of reactive oxygen species and subsequent activation of NFκB associated with transcription activation of a host of proinflammatory mediators.19 In addition, peroxynitrite is known to modulate cell signaling by both phosphorylation as well as oxidation of critical protein tyrosine residues along the mitogen-activated protein kinase pathways.20
Mitochondria
It has been estimated that more than 1 billion years ago some aerobic bacteria invaded and subsequently colonized some form of primordial eukaryotic cells that themselves lacked the ability to use oxygen. At some point a symbiotic relationship developed between the cell and the aerobic bacteria, and this relationship has remained steadfast through evolution.21,22 Evidence for the persistence of this endosymbiotic relationship suggesting that mitochondria have retained many aspects of their bacterial origins includes the observations that mitochondria only arise from other mitochondria; that mitochondria maintain their own genome; that the mitochondrial chromosome is bacteria-like, circular in structure, and without associate histones; that mitochondria synthesize their own proteins; and that these mitochondrial proteins, like bacterial proteins, exhibit N-formyl methionine; and that antibiotics that inhibit protein synthesis in bacteria are also toxic to mitochondria.23
Mitochondria are responsible for the generation of more than 95% of ATP synthesized to support aerobic respiration (Figure 74-3). Enormous oxygen consumption by “power plant” mitochondria represents a double-edged sword, on the one hand permitting efficient production of ATP, but on the other hand a potential source of high quantities of reactive oxygen species. Assuming 388 L of oxygen consumed per day by a normal adult, this would require 2 × 1019 molecules of cytochrome oxidase. Considering this requirement as well as that of other members of the mitochondrial electron transport chain outlined below, the surface area of mitochondrial cristae inner membrane to scaffold this quantity of respiration elements24 is approximately 14,000 m2.
In addition to their role in facilitating aerobic respiration, mitochondria are also involved in intracellular signaling, intracellular calcium regulation,25 cellular differentiation and growth, and cellular death pathways.26,27 With respect to the latter function, the mitochondria provide close monitoring for a number of cellular “danger signals.” These include loss of transmembrane potential, decreased ATP production, increased influx of reactive oxygen and reactive nitrogen species, leakage of mitochondrial proteins into the cytoplasm via mitochondrial permeability transition pores, activation of hypoxia-responsive genes, and recognition by the cell of mitochondrial proteins with signature N-formyl methionine.23 Mitochondria in cells may undergo cycles of both fusion and fission.28 If the mitochondria “senses” overwhelming cell damage as manifested by excessive reactive oxygen species, DNA damage, denatured protein, ongoing inflammation, hypoxia, or even deprivation of growth factors, the mitochondrial pathway of cellular apoptosis may be initiated.29,30 If apoptosis is initiated by excessive reactive oxygen species, activation of the sphingomyelin/ceramide cycle precedes apoptosis.
Critical mitochondrial damage results in increased mitochondrial permeability, the mitochondrial permeability transition that results in release of proapoptotic proteins, including cytochrome c from the mitochondria into the cytoplasm. This results in activation of various caspase cascades and resultant DNAase activation and DNA dissolution.31 Activation of the apoptotic pathway orchestrating cell death is also governed by the relative concentrations of proteins BCL-2 and BCL-X. Integrity of the mitochondrial transport chain complexes defines a fine line between maintaining mitochondrial homeostasis and efficient respiration versus excessive oxygen free radical production, mitochondrial dysfunction, and mitoptosis. This balance is maintained through multiple feedback signals related to mitochondrial proton and calcium concentrations, reactive oxygen species, overall mitochondrial redox state, mitochondrial membrane potential, and mitochondrial matrix pH, all of which affect the cascade of electron transfer along the mitochondrial transport chain.23 It has been demonstrated that pathologic metabolic downregulation of cytochrome oxidase, such as in the setting of excessive mitochondrial production of reactive nitrogen and oxygen species, manifests as multiple organ dysfunction syndrome.32
Alterations in mitochondrial function in the setting of shock have been extensively scrutinized.33–35 In addition to decreased activity of specific mitochondrial enzymes previously indicated, altered translocase activity and mitochondrial calcium transport are also coincident with actual changes in mitochondrial morphology. In sepsis, increased NO production has been associated with activation of the poly-adenosine ribosyl phosphorylase (PARP) pathway, the former associated with impaired binding of oxygen to cytochrome oxidase and the latter associated with cellular depletion of NADH.36 In various models of multiple organ dysfunction syndrome and sepsis, mitochondrial permeability transition has been shown to result in collapse of the mitochondrial electrochemical gradient and impairment of ATP synthesis coincident with calcium influx into the mitochondria. Sepsis inflammation can induce a vicious cycle of oxidative mitochondrial damage leading to an increased flux of reactive oxygen species that further damage mitochondrial respiration components, resulting in mitochondrial glutathione depletion, enhanced mitochondrial membrane lipid peroxidation and decreased copy number, increased deletions, and decreased messenger RNA transcripts from mitochondrial DNA (Figure 74-4).37
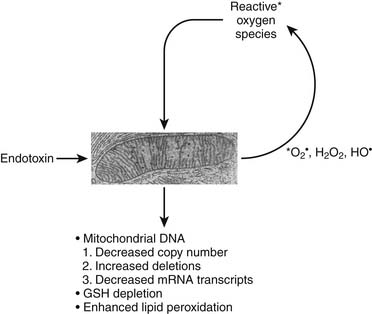
Figure 74–4 Vicious cycle mitochondrial injury in the setting of excessive reactive nitrogen/oxygen species.
The independent mitochondrial genome; located within the mitochondrial matrix, is comprised of double-stranded, circular DNA of 16.6 kb and codes for 37 genes, including proteins involved in various redox reactions, oxidative phosphorylation, ATP synthesis, and enzymes comprising the Krebs cycle, fatty acid beta-oxidation, and pyruvate oxidation.38 Mitochondrial DNA mutations associated with the gene coding for cytochrome oxidase have been clinically linked to Leigh syndrome; mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke (MELAS); encephalomyopathy; lactic acidosis; and mitochondrial proliferation in muscle biopsies, the so-called characteristic histopathology of ragged red fibers.21
Adenosine Triphosphate
ATP is considered the molecular unit of intracellular energy currency. ATP derives its inherent energy secondary to anhydride bonds connecting adjacent phosphate functional groups. Hydrolysis of ATP energy generates energy for all cellular processes. In addition ATP also serves as a cofactor for signal transduction reactions using a variety of kinases as well as adenyl cyclase. Normally cellular ATP concentration is maintained in the range of 1 to 10 mmol/L, with a normal ratio of ATP/ADP of approximately 1000. Totally quantity of ATP in an adult is approximately 0.10 mol/L. Approximately 100 to 150 mol/L of ATP are required daily, which means that each ATP molecule is recycled some 1000 to 1500 times per day. Basically, the human body turns over its weight in ATP daily.39 Transmembrane proton flux through the mitochondrial ATPase synthase complex occurs at an estimated rate of 3 × 1021 protons per second. This corresponds to ATP reformed at a rate of 9 × 1020 molecules/sec, or approximately 65 kg ATP recycled per day in a normal resting adult (Figure 74-5).24
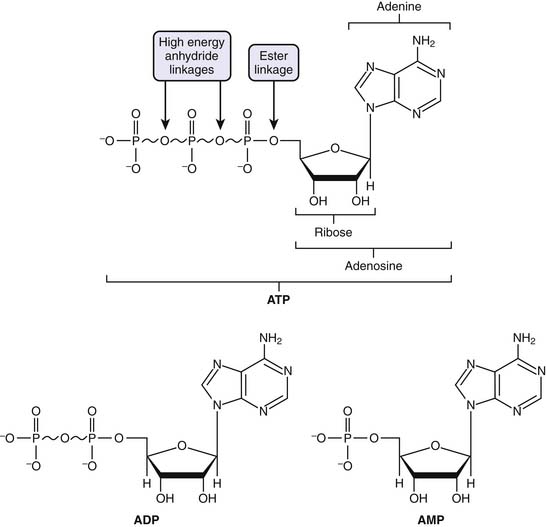
Respiration, Metabolic Pathways
Glycolysis
Acetate coupled to coenzyme A (AcCoA) is derived from carbohydrates, lipids, and proteins. Glucose is transported into cells via glucose transporter (GLUT) receptors and osmotic gradients (see Chapter 77). Ten enzymatic reactions within the cell cytoplasm define the metabolic pathway, termed glycolysis. These initial series of reactions ultimately generate two net molecules of ATP, two molecules of NADH, and two molecules of pyruvate.
The fates of pyruvate is multiple. Under anaerobic conditions, pyruvate may be reduced by NADH to lactate to regenerate NAD+. Alternatively, pyruvate is shuttled to the mitochondria, where it is further metabolized to carbon dioxide (CO2) and AcCoA. Pyruvate transamination yields alanine, whereas pyruvate carboxylase generates oxylacetate (Figure 74-6).
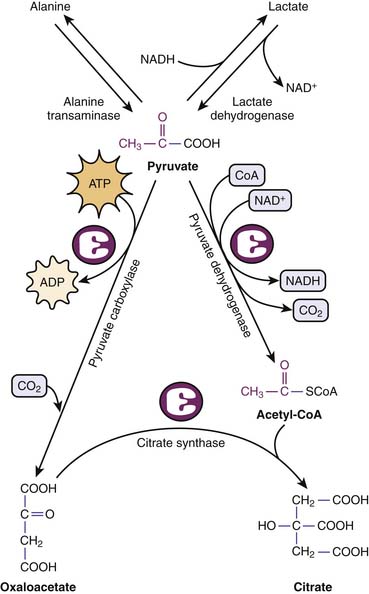
Figure 74–6 Metabolic fates of pyruvate, end product of glycolysis.
(Modified from Baynes JW, Dominiczak MH: Medical biochemistry, New York, 2009, Mosby Elsevier.)
Catabolism of fatty acids by β-oxidation generates one molecule of AcCoA and one molecule each of FADH2 and NADH for each two-carbon fatty acid fragment cycle. These reactions occur in the mitochondria after fatty acid transport by a carnitine transport system. It should be appreciated that generation of AcCoA by fatty acid β-oxidation occurs independent of pyruvate dehydrogenase that can be rate limiting for complete glucose metabolism. Particularly as an aspect of the metabolic stress response mediated by cortisol, catecholamines, and interleukins 6 and 2, protein degradation can occur with release of amino acids. All amino acids may be catabolized to either AcCoA or some Krebs cycle intermediate. Accordingly, amino acids can be mobilized for energy production as well as de novo protein synthesis. Alternatively, amino acids can undergo gluconeogenesis, a costly process that basically requires four ATP molecules plus two GTP molecules and two NADH molecules to regenerate one molecule of glucose from two molecules of pyruvate (see Chapter 77). ATP and GTP for these series of reactions are provided by β-oxidation of fatty acids.
Krebs Cycle
The Krebs cycle summarizes a circular series of reactions in the mitochondria to metabolize AcCoA to two molecules of CO2 with resultant generation of one molecule of GTP, three molecules of NADH, and one molecule of FADH2. GTP is equivalent to ATP in terms of energy charge. Although oxygen itself is not part of the Krebs cycle, its presence at the end of the mitochondrial electron transport chains ensures recycling of NAD+ and FAD required in the Krebs cycle (Figure 74-7).
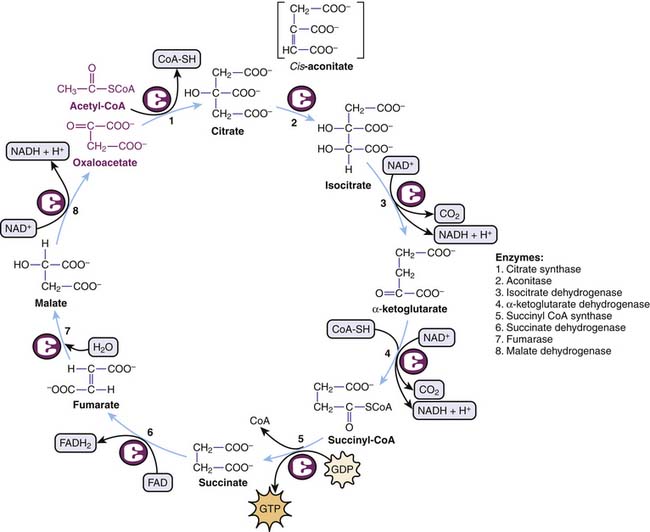
Figure 74–7 Circular series of reactions involved in the Krebs cycle.
(Modified from Baynes JW, Dominiczak MH: Medical biochemistry, New York, 2009, Mosby Elsevier.)
Electron Transport Chain
Electrons derived from reducing equivalents NADH and FADH2 are shuttled along the mitochondrial electron transport chain, ultimately reducing molecular oxygen to water. Basically, for each pair of electrons involved in one hydride equivalent, three molecules of ATP are produced. Five complexes of proteins and cytochromes comprise the mitochondrial electron transport chain and facilitate a step-down flow of FADH2 and NADH reduction potential along the inner membrane of the mitochondria. These redox reaction complexes include NADH dehydrogenase–ubiquinone oxidoreductase (complex 1), succinate dehydrogenase–ubiquinone oxidoreductase (complex 2), ubiquinone–cytochrome C oxidoreductase (complex 3), cytochrome c oxidase (complex 4), and ATP synthase (complex 5). Electrons in the form of hydride ions from NADH and FADH2 cascade along these protein/cytochrome complexes toward molecular oxygen. Protons generated during these reactions are pumped across the inner mitochondrial membrane matrix into the intermembrane space, generating a proton motive force. The proton electrochemical gradient across the inner mitochondrial membrane drives ATP synthesis by a reaction that has been termed chemiosmosis.41 Energy for ATP synthesis arises from an influx of these protons back into the matrix, literally through the rotary motor of ATP synthase.42
The proton pore involves the c-ring and the a-protein. The rotary component is the coiled-coil γ-subunit, which is bound to the ε-subunit and to the c-ring. The stationary component is the hexameric α3β3 unit and is fixed by the δ, b, and a proteins (Figure 74-8).
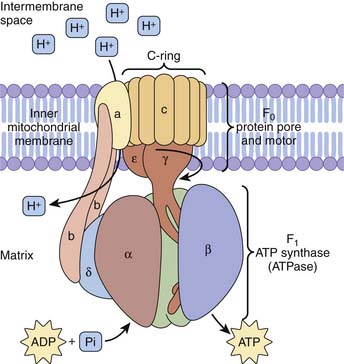
Figure 74–8 ATP synthase complex consists of a motor (proton gradient pore, F0) and generator (ATP synthase, F1).
(Modified from Baynes JW, Dominiczak MH: Medical biochemistry, New York, 2009, Mosby Elsevier.)
Monitoring of Tissue Oxygenation
Blood Lactate Monitoring in Critically Ill Patients
In general, lactic acidosis occurs when there is an imbalance between production and clearance of lactate. Measurement of lactate in human blood was first described in 1843 in a lethal case of fulminant septic shock due to puerperal fever in a young woman.43 Blood lactate monitoring is frequently performed in critically ill patients, usually with the aim of detecting tissue hypoxia44 with resultant anaerobic hyperlactatemia, traditionally termed type A lactic acidosis. However, other processes not related to tissue hypoxia can also result in increased blood lactate levels,45 such as aerobic hyperlactatemia or type B lactic acidosis that complicates clinical interpretation and therapy in cases of elevated lactate concentration. Although lactic acidosis is often associated with a high anion gap and is generally defined as a lactate level greater than 5 mmol/L and a serum pH less than 7.35, the presence of hypoalbuminemia may mask the anion gap and concomitant alkalosis may raise the pH. As originally reported by Huckabee,45a normal arterial blood lactate is approximately 0.620 mmol/L and venous lactate is slightly higher at 0.997 mmol/L.
Anaerobic Hyperlactatemia
During anaerobic conditions, pyruvate derived from the conversion of glucose cannot enter the Krebs cycle via AcCoA to produce energy. Instead, pyruvate is converted into lactate, known as the Pasteur effect. The normal lactate/pyruvate ratio is approximately 20:1, and it rises under hypoxic conditions. The causal relationship between anaerobic hyperlactatemia and tissue hypoxemia has been confirmed by experimental46,47 and clinical44 studies; decreasing systemic oxygen delivery (DO2) until oxygen demand can no longer be met and limiting oxygen consumption by DO2 coincides with a sharp increase in lactate levels. Accordingly, in the early phase of septic shock hyperlactatemia is accompanied by oxygen supply dependency.48 In severe sepsis or septic shock prior to resuscitation, hyperlactatemia coincides with a low central venous oxygen saturation49 and increased lactate/pyruvate ratios,50 whereas increases in DO2 are associated with reductions in lactate.49 No critical level of DO2 or SvO2 could be associated with hyperlactatemia. This could represent regional differences in DO2 and demand.51 However, improving capillary perfusion was shown to correlate with a reduction in lactate levels in patients with septic shock, independent of changes in systemic hemodynamic variables.52 The latter observation illustrates the hypothesis that, in the absence of low systemic DO2 relative to systemic metabolic demand, microcirculatory processes hampering oxygen use at the tissue level may raise lactate levels.
Aerobic Hyperlactatemia
Various studies demonstrate that mechanisms other than tissue hypoxia can account for hyperlactatemia, including (1) mitochondrial dysfunction35,53,54; (2) decreased lactate clearance in liver dysfunction,55,56 status post liver surgery57 and cardiac surgery,58 as well as in sepsis,59,60 where it was shown to predict poor outcome61; (3) increased aerobic glycolysis resulting in amounts of pyruvate exceeding the pyruvate dehydrogenase capacity that can be triggered by cytokine-mediated uptake of glucose62,63 or catecholamine-stimulated increased Na-K pump activity45,64–67; (4) impaired activity of pyruvate dehydrogenase, essential for the conversion of pyruvate into AcCoA, that can be inhibited in sepsis,68,69 as well as in thiamin deficiency (beriberi)70; (5) alkalosis71 by increasing cellular lactate efflux due to an H+-linked lactate carrier mechanism across the cell membrane; (6) acute lung disease55,72; (7) several drugs and intoxications such as epinephrine (by increased glycogenolysis, glycolysis, and stimulation of the Na-K pump),73,74 metformin (particularly in the presence of renal insufficiency), and nucleosidic reverse transcriptase inhibitors for the treatment of HIV (by inducing mitochondrial cytopathy)75–77; and (8) intoxications with methanol, cyanide (by inhibition of oxidative phosphorylation),78 or ethylene glycol (by artifactual reaction of lactate electrodes).79
In summary, during critical illness, the source of lactate is often assumed to be ischemic tissues that use anaerobic metabolism. This is superficially supported by lactate as an adverse prognostic marker.80,81 However, lactate metabolism in critical illness is complex and often does not indicate ischemic tissues.82,83 A common clinical scenario occurs during volume resuscitation of patients (iatrogenic hyperchloremia) or epinephrine infusions, in which a chloride load and type B lactic acidosis are interpreted by the body as “shock,” requiring more fluid and more vasoactive-inotropic support. This scenario may initiate a vicious cycle and potential over-resuscitation.84 It is essential to diagnose this syndrome correctly and adjust management accordingly.
Given the available evidence, patients presenting with hypotension and an elevated lactate level (>5 mmol/L) have a grave prognosis with a high mortality rate (>80%). Likewise, patients who are critically ill and do not clear lactate by 48 hours have a similarly high mortality rate.85 There are no data currently available, however, to suggest that monitoring lactate levels beyond this time frame is prognostically useful. Early goal-directed therapy in severe sepsis is an example of outcome benefit that arguably targets the patient’s primary pathophysiology rather than lactic acidosis per se.49,86 Hence, there are no current data demonstrating that therapeutic maneuvers specifically targeted at decreasing lactate levels are beneficial. The treatment of the patient with lactic acidosis should therefore be aimed at the underlying disease and the maintenance of organ perfusion and not the lactate concentration itself. Accordingly, the routine daily measurement of serum lactate levels in critically ill patients remains controversial.
Continuous Central Venous Oxygen Saturation Monitoring
Monitoring of the mixed venous oxygen saturation (SvO2) is used as a surrogate for quantifying the balance between systemic oxygen delivery and consumption during the treatment of critically ill patients.87 Measurement of SvO2 requires placement of a pulmonary artery catheter with a risk/benefit ratio that is still a matter of controversy in adult patients88–90 and rarely performed in pediatric patients. However, measuring central venous oxygen saturation (ScvO2) only requires a central venous catheter, routinely inserted in critically ill patients, and a fiber optic catheter with a spectrophotometer for continuous ScvO2 assessment. ScvO2 largely reflects the degree of oxygen extraction from the brain and the upper part of the body since the catheter tip usually is located in the superior vena cava. At baseline, ScvO2 is about 2% to 3% less than SvO2 because, overall, the lower body extracts less oxygen than the upper body, resulting in a higher inferior vena cava oxygen saturation. Readings may be taken intermittently by blood sampling and co-oximetry or continuously with a fiberoptic catheter. However, beneficial effects on patient outcome to date49 have only been demonstrated with continuous measurement of ScvO2.
Low values of SvO2 or ScvO2 indicate a mismatch between oxygen delivery and tissue oxygen need.91 Monitoring ScvO2 has been successfully used as a hemodynamic goal in the management of early sepsis. Rivers et al.49 demonstrated in a prospective randomized study in adult patients with severe sepsis and septic shock that, in addition to maintaining central venous pressure in the range of 8 to 12 mm Hg, mean arterial pressure above 65 mm Hg, and urine output higher than 0.5 mL/kg/hr, maintenance of an ScvO2 above 70% resulted in an absolute reduction of mortality by 15%. Thus the guidelines of the Surviving Sepsis Campaign92 stated that the use of SvO2 and ScvO2 is equivalent in the management of adult patients with severe sepsis and septic shock. In pediatric patients with septic shock, directing treatment to the end point of ScvO2 at 70% or greater was also associated with a significant decrease in the 28-day mortality rate from 39.2% to 11.8%.93 ScvO2 goal-directed therapy resulted in more crystalloid, blood product, and inotropic support during the first 6 hours compared with the control group receiving standard resuscitation.
Continuous monitoring of SvO2 is interesting, but clear indications for this technique in children as well as ScvO2 target values remain to be determined. It has been shown that exact numeric values of SvO2 and ScvO2 saturations are not equivalent in various hemodynamic conditions.94 Some authors have therefore argued that ScvO2 cannot be used as surrogate for SvO2 under conditions of circulatory shock.95 However, for clinical purposes, the trends between these values were found to be reliable and clinically valuable.96 Based on the evidence available, the primary indication for the use of central venous oximetry is as part of the early resuscitation in severe septic and septic shock patients. Further research is needed to elucidate the efficacy of venous oximetry in other patient groups as well as to assess whether an intermittent measurement using central venous blood gas analysis is as effective as continuous measurement of ScvO2 using fiberoptic catheters.
Functional Magnetic Resonance Imaging
Functional magnetic resonance imaging (fMRI) is a noninvasive technique for measuring brain activity. It works by detecting the changes in blood oxygenation and flow that occur in response to neural activity. When a brain area is more active, it consumes more oxygen; to meet this increased demand, blood flow increases to the active area.97,98 Hemoglobin is diamagnetic when oxygenated but paramagnetic when deoxygenated. This difference in magnetic properties leads to small differences in the MR signal of blood depending on the degree of oxygenation. Since blood oxygenation varies according to the levels of neural activity, these differences can be used to detect brain activity. This form of MRI is known as blood oxygenation level–dependent (BOLD) imaging, which is the most widely used method of fMRI with T2∗-weighted imaging revealing changes in vascular oxygenation. A limitation is that the technique is also sensitive to changes in hemoglobin concentration that may result from alterations in vascular volume and flow as well as interconversion of oxyhemoglobin and deoxyhemoglobin. Therefore this technique provides qualitative assessment of changes in oxygenation rather than quantitative measurements. Thought to primarily reflect changes in blood flow, BOLD is widely used for functional brain mapping.99–101 BOLD is starting to be applied to tumor studies.102–104 BOLD is particularly responsive to oxygen manipulation accompanying hyperoxic gas breathing as a simple way to ameliorate hypoxia.105,106 This technique may also allow direct estimates of PO2, such as in the superior mesenteric vein or heart of children. A recent study in pediatric patients with complex congenital heart disease showed a strong correlation between oxygen saturations measured in the cardiac catheterization lab and saturations obtained noninvasively by BOLD MRI.107 However, fMRI remains difficult to perform in critically ill unstable patients; consequently, few teams have acquired the equipment and experience necessary to apply this technique.108
Magnetic Resonance Spectroscopy
MRI provides anatomic images and morphometric characterization of disease, whereas magnetic resonance spectroscopy (MRS) provides noninvasive metabolite/biochemical information about tissues in vivo. MRS has been used clinically for more than 2 decades. The major applications of this advanced tool include investigation of neurologic and neurosurgical disorders, including blood vessel distribution and architecture, blood flow velocity, regional perfusion and blood volume, blood and tissue oxygenation, lactate production and intracellular pH, Krebs cycle activity, and mitochondrial oxidative phosphorylation.109 Phosphorus-31 MRS detects compounds involved in energy metabolism such as creatine phosphate, ATP and inorganic phosphate, and certain compounds related to membrane synthesis and degradation. Proton MRS is most commonly used. Four main markers related to O2 metabolism are studied: the peak of N-acetyl-aspartate (NAA), an amino acid present in neurons that reflects the status of neuronal tissue; creatine, found in glia and neurons, that serves as a point of reference because its level is believed to be stable; choline, a constitutive component of cell membranes that reflects glial proliferation or membrane breakdown110; and lactate, a marker of anaerobic metabolism and therefore of ischemia.111 Elevated lactate and decreased NAA are associated with worse neurologic outcome in neonates with asphyxial injury.112,113 Increased lactate may persist up to 1 week after asphyxial injury.114 Compared with positron emission tomography (PET), which is frequently used to evaluate tumor hypoxia but requires injections of radioactive isotopes, multiple acquisitions, and, therefore, extended imaging times, fMRI and MRS have a number of advantages. Both MRI and MRS are currently used primarily for outcome prediction and not minute-to-minute patient management.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


