148 Cardiovascular Drugs
 Key Points
Key Points• Cardiovascular drugs are responsible for many fatalities.
• β-Receptor antagonists, calcium channel antagonists, and digoxin primarily cause toxicity by disruption of intracellular calcium homeostasis and lead to hypotension and dysrhythmias.
• With sustained-release forms of calcium channel antagonists and beta-receptor antagonists, toxicity has a delayed peak and longer duration, which can lead to cardiovascular collapse and arrest if treatment is delayed or there is insufficient cardiovascular moritoring.
• Diagnostic testing should include continuous cardiac monitoring; electrocardiogram; measurement of appropriate serum drug concentrations, electrolytes, and glucose; and investigation of coingestants.
• The call to the pharmacist to obtain the hyperinsulinemia-euglycemia infusion in calcium channel antagonist overdose should be made when the norepinephrine infusion is begun.
Epidemiology
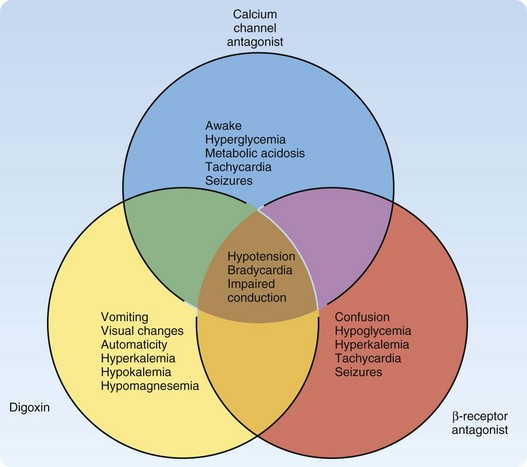
The 2009 Annual Report of the American Association of Poison Control Centers National Poison Center Database System reported cardiovascular drugs as the second most common cause of fatalities overall (10%), and they were the second fastest in rate of exposure increase.1 Cardiovascular drugs as a category were ranked as the fifth leading cause of death (44 total deaths: 5 from β-receptor antagonists, 16 from calcium channel antagonists, and 23 from cardiac glycosides). These specific cardiovascular drugs share the clinical effects of hypotension, bradycardia, and conduction disturbances. However, unique differences can help distinguish them in an unknown overdose (Fig. 148.1). Other pharmaceuticals included in the category of cardiovascular agents are angiotensin-converting enzyme inhibitors, antiarrhythmics, clonidine, and other antihypertensives; they are not discussed in this chapter.
Calcium Channel Antagonists
Pathophysiology
Calcium channel antagonists block the intracellular flow of calcium ions through L-type voltage-gated calcium channels in myocardial, smooth muscle, and pancreatic beta-islet cells. These mechanisms of action result in cardiovascular toxicity both directly and indirectly. Depending on the selectivity of the calcium channel antagonist, the direct cardiovascular toxicity is a combination of the effects on the cardiac conduction system, myocardial contractility, and vascular smooth muscle vasodilation. The dihydropyridine class (e.g., amlodipine, nifedipine) preferentially acts on the peripheral vasculature, thereby potentially leading to hypotension and reflex tachycardia. Verapamil operates on the sinoatrial and atrioventricular (AV) nodes and on the myocardium. Diltiazem acts to a lesser extent than verapamil on the cardiac tissue and nodes, and it also dilates peripheral vasculature (Table 148.1). The degree of contribution from each mechanism of cardiovascular toxicity can be difficult to predict. Despite the differences in therapeutic mechanisms, the distinctions among families of calcium channel antagonists are often blurred during an overdose, and the patient generally suffers from negative chronotropic, inotropic, and dromotropic effects.2
Table 148.1 Classification of Calcium Channel Antagonists
| CLASS | ACTION(S) | EXAMPLE(S) |
|---|---|---|
| Phenylalkylamines | Act on sinoatrial and atrioventricular nodes and the myocardium | Verapamil (Calan) |
| Benzothiazepines | Dilate peripheral vasculature and act to a lesser degree than verapamil on cardiac tissues and nodes | Diltiazem (Cardizem, Tiazac) |
| Dihydropyridines | Act on peripheral vasculature, leading to hypotension and reflex tachycardia |
Presenting Signs and Symptoms
Verapamil and diltiazem overdoses cause bradycardia and numerous conduction abnormalities in and below the AV node. The decreased blood pressure results from vasodilation and decreased cardiac contractility from the negative inotropic and chronotropic effects.2
Differential Diagnosis and Medical Decision Making
The differential diagnosis (see also Fig. 148.1) for overdose of calcium channel antagonists includes other cardiovascular drugs such as beta-blockers, clonidine, digitalis, and other antidysrhythmics. The emergency physician should also consider myocardial infarction and other causes of cardiogenic shock. The potency of the effect of calcium channel antagonists on the cardiovascular system is astounding. Significant cardiovascular toxicity can occur after supratherapeutic ingestion of calcium channel antagonists. Ingestion of double the therapeutic dose should instigate medical evaluation and treatment. Immediate-release calcium channel antagonists should have some clinical effect within 6 hours. Sustained-release calcium channel antagonists should result in clinical manifestations within 1 to 14 hours.3
Diagnostic testing is contingent on the necessity of treatment for hemodynamic instability. Once the patient’s airway, breathing, and cardiovascular status have been assessed and stabilized, testing should start with a 12-lead electrocardiogram (ECG) and chest radiography. Rapid determination of hyperglycemia and metabolic acidosis with capillary glucose and arterial blood gas analysis may demonstrate a severe calcium channel antagonist overdose. An elevated serum lactate concentration may be another marker of severe calcium channel antagonist overdose.4 Testing for serum concentrations of calcium channel antagonists is not clinically useful or available to guide treatment. Otherwise, standard laboratory testing for a general overdose is a good comprehensive approach.
Treatment
Whole-bowel irrigation has been suggested for overdose of calcium channel antagonists because many of these drugs are sustained-release preparations. Whole-bowel irrigation is not indicated for a patient with hemodynamic instability because a significant amount of the drug has already been absorbed,5 and, therefore, the opportunity for prevention has passed. In addition, challenging a hypoperfused gastrointestinal system can have disastrous consequences, such as functional and physical obstruction by a calcium channel antagonist bezoar,6–9 as well as perforation. Generally speaking, no evidence indicates that any gastric decontamination procedure improves outcome in the patient with an overdose, and the risks must be assessed against the benefits.
An antidotal treatment regimen is provided in Figure 148.2. This regimen emphasizes elemental calcium, either as calcium gluconate (30 mL of a 10% solution, or 3 g of calcium gluconate; 14 mEq elemental calcium) or calcium chloride (10 mL of a 10% solution, or 1 g; 13.5 mEq of elemental calcium). Calcium chloride should be administered through central venous access because it is an acidifying salt, which could cause necrosis of the peripheral vasculature. If the intravenous calcium boluses appear to have improved hemodynamic status, close monitoring for recrudescence of toxicity must be maintained, and further boluses must be given as necessary. An intravenous infusion of calcium is warranted only when it effectively treats the hypotension, and further boluses are required to support the blood pressure (Table 148.2). The serum calcium concentration should be monitored, but antidotal treatment rarely gives rise to clinically significant hypercalcemia.
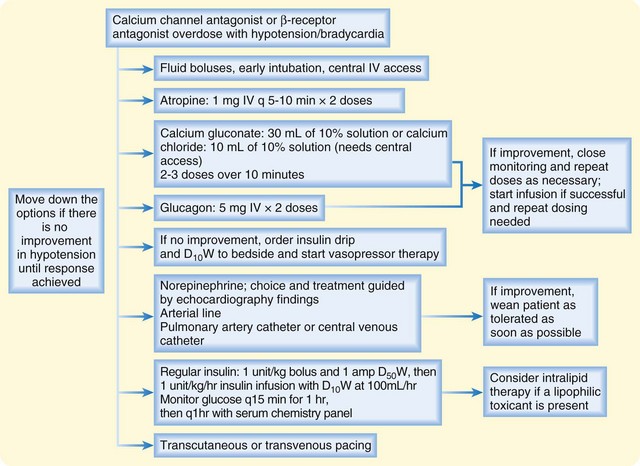
Fig. 148.2 Treatment of overdose with calcium channel antagonist or β-receptor antagonist.
D10W, 10% dextrose in water; D50W, 50% dextrose in water; IV, intravenous(ly).
Table 148.2 Antidotes, Treatments, Facts, and Formulas for Cardiovascular Drugs
| ANTIDOTE OR TREATMENT | DOSING | ADVERSE EFFECTS and signs of improving perfusion |
|---|---|---|
| Atropine | Anticholinergic toxicity | |
| Glucagon | ||
| Calcium chloride (10 mL of 10% solution = 1 g = 13.5 mEq elemental calcium) | ||
| Calcium gluconate (10 mL of 10% solution = 1 g = 4.65 mEq elemental calcium) | Hypercalcemia | |
| Norepinephrine (α1 and β1 agonist) | Start at 0.1 mcg/kg/min; titrate to MAP of 70 mm hg and improvement in perfusion | |
| Dopamine | Same as for norepinephrine | |
| Epinephrine (α1, β1, and β2 agonist) | Start at 1 mcg/min; titrate to effect | Same as for norepinephrine |
| Dobutamine (β1 agonist) | 2.5 mcg/kg/min to 15 mcg/kg/min | Same as for norepinephrine |
| Isoproterenol (β1, β2 agonist) | — | |
| Insulin (regular) | Consultation with a clinical toxicologist is recommended Give a bolus of 1.0 units/kg IV with 1 ampule of D50W Immediately start an infusion of 1.0 units/kg/hr with a glucose infusion of D10W at 100 mL/hr Titrate 0.5 units/kg every 30 min until desired effect; 1 ampule of D50W can be given during every increase in infusion; titrate to desired effect when blood pressure has reached desired value and signs of improving perfusion When the blood pressure has reached the desired value with the insulin infusion, taper and wean pressor agent therapy Monitor serum glucose concentration every 15 min for the first 60 min; when stable, monitor every 60 min thereafter Monitor serum potassium and other electrolyte concentrations every 60 min | |
| Intralipid 20% | ||
| Intravenous crystalloid | 20 mL/kg IV; repeat again if blood pressure has not improved | Pulmonary edema with severe cardiogenic shock |
| Vasopressin (V1, V2 receptor agonist) | 0.01-0.04 units/min; titrate to effect along with administration of 1-2 catecholamine vasopressors | |
| Phosphodiesterase inhibitor (Milrinone) | Give 50 mcg/kg IV bolus over 2 min; then 1.0 mcg/kg/min | — |
| Digoxin-specific Fab fragments | For acute overdose (unknown amount in adult or child): 5-10 vials IV as a bolus; repeat as needed every 30 min For chronic overdose in adult or child: 1-2 vials IV as a bolus; repeat as needed every 30 min If amount ingested is known: 1 vial binds 0.5 mg of digoxin Dosage calculated from serum digoxin concentration:
| |
| Mechanical devices | — |
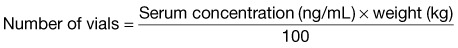
D10W, 10% dextrose in water; D50W, 50% dextrose in water; IV, intravenous(ly); MAP, mean arterial pressure.
Hyperinsulinemia euglycemia (HIE) therapy and catecholamines with inotropic and vasopressor activity are the next line of treatment for refractory hypotension in calcium channel antagonist overdose, but inotropics and vasopressors will be discussed first. A multicenter study compared dopamine and norepinephrine agents in all patients categorized as being in shock, regardless of cause. No difference was seen in the outcome (death at 28 days) for all patients in the study, but a predetermined subgroup analysis found greater mortality in patients with cardiogenic shock who were treated with dopamine.10 Because calcium channel antagonists can cause cardiogenic shock, norepinephrine is probably a good first choice. If clinically significant hypotension persists, adding more agents may be necessary. Cardiovascular data from diagnostic modalities such as transthoracic echocardiogram, pulmonary artery catheter, arterial catheter, and central venous catheter should dictate which cardiovascular agent is the most appropriate choice. Vasopressin has been used in human cases when peripheral vasoconstriction is indicated.11 Worsening of the cardiac index was demonstrated when vasopressin was used in an animal model to treat hypotension induced by calcium channel antagonist.12
In 1999, Yuan et al.13 described the first published use of high-dose insulin-euglycemia (HIE) therapy, in four patients with verapamil overdose and in one patient with amlodipine and atenolol overdose. HIE promotes inotropy by improving myocardial energy production. In addition, insulin has antiinflammatory attributes that protect against apoptosis and ischemic reperfusion injury.2
Subsequently, numerous case reports, reviews, and HIE regimens were published.2,14–19 HIE therapy has successfully reversed cardiogenic shock from a polydrug overdose,20 and historically it was used in multiple nontoxicologic conditions, such as acute myocardial infarction, post–cardiac surgery status, and septic shock.21 The superiority of HIE therapy for cardiogenic shock resulting from calcium channel antagonist toxicity was also demonstrated dramatically in animal models.22–25
Failure of HIE therapy often occurs when it is started as rescue therapy and when the dose is inadequate.26 A small prospective observational study of patients treated with HIE therapy supports it safety.27 HIE therapy should be started when boluses of calcium, glucagon, atropine, and intravenous fluids have failed and the physician is considering a pressor agent to improve refractory hypotension. The call to the pharmacist to obtain the HIE infusion should be made when the norepinephrine infusion is begun.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


