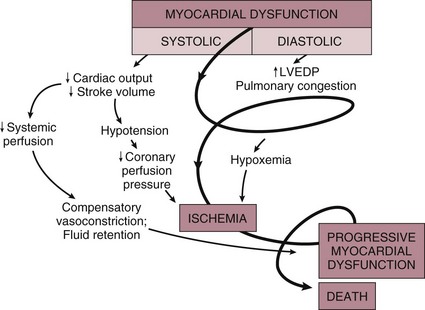
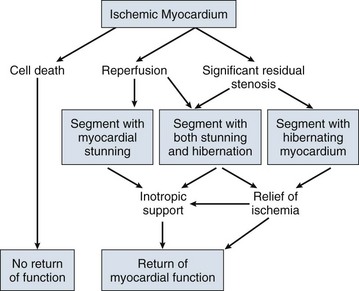
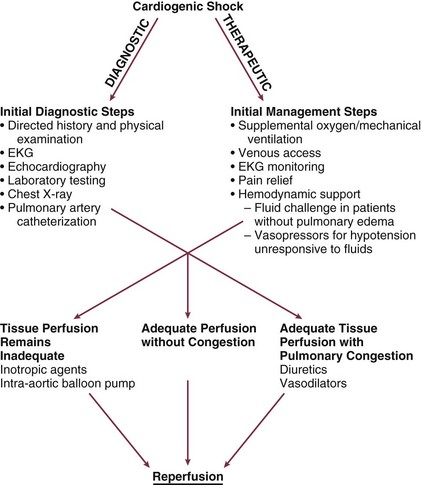
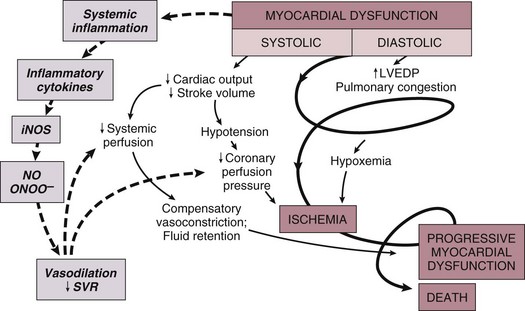
22 Cardiogenic shock is the syndrome that ensues when the heart is unable to deliver enough blood to maintain adequate tissue perfusion. Acute myocardial infarction (MI) is the leading cause, but other potential etiologic factors need to be considered.1,2 Without prompt diagnosis and appropriate management, morbidity and mortality rates are substantial, approaching 60% for all age groups.2,3 Rapid evaluation and prompt initiation of supportive measures and definitive therapy in patients with cardiogenic shock may improve both early and long-term outcomes. The clinical definition of cardiogenic shock includes decreased cardiac output and evidence of tissue hypoxia in the presence of adequate intravascular volume. The diagnosis of circulatory shock (Box 22.1) is made at the bedside by the presence of hypotension along with a combination of clinical signs indicative of poor tissue perfusion, including oliguria, clouded sensorium, and cool, mottled extremities. Hemodynamic criteria include sustained hypotension (systolic blood pressure < 90 mm Hg for at least 30 minutes) and a reduced cardiac index (<2.2 L/min/m2) in the presence of elevated filling pressures (pulmonary capillary occlusion pressure > 15 mm Hg).4 Cardiogenic shock is diagnosed after documentation of myocardial dysfunction and exclusion or correction of factors such as hypovolemia, hypoxia, and acidosis. Pump failure due to cardiogenic shock has long been known to carry a high mortality rate. The seminal article outlining prognosis after MI was a single center series of 250 patients reported by Killip in 1967.5 Killip divided patients into four classes as follows: Killip class I: no evidence of congestive heart failure Killip class II: presence of an S3 gallop and/or bibasilar rales Killip class III: pulmonary edema (rales greater than halfway up the lung fields) Nineteen percent of the 250 patients were in class IV at presentation, and their mortality rate was 81%.5 With the advent of right-sided heart catheterization, Forrester and Swan defined hemodynamic subsets after MI analogous to the clinical subsets outlined by Killip.4 Subset I consisted of patients with normal pulmonary capillary wedge pressure (PCWP) and cardiac output, subset II consisted of patients with elevated PCWP and normal cardiac output, subset III consisted of patients with normal PCWP and decreased cardiac output, and subset IV consisted of patients with elevated PCWP and decreased cardiac output.4 Despite advances in management of heart failure and acute MI, the mortality rate of patients with cardiogenic shock has remained high.2,6–8 Data suggest an increase in survival in the 1990s, coincident with the use of reperfusion strategies.7–9 Cardiogenic shock, however, remains the most common cause of death in hospitalized patients with acute MI. Accurate determination of the precise incidence of cardiogenic shock is difficult because patients who die of MI prior to reaching the hospital generally do not receive this diagnosis.6,10–13 Nonetheless, estimates from a variety of sources have been fairly consistent. The Worcester Heart Attack Study,6 a community-wide analysis, found an incidence of cardiogenic shock of 7.5%, an incidence that remained fairly stable from 1975 to 1997.6,9 The incidence was similar in the randomized GUSTO (Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries) trial (7.2%),14 in other multicenter thrombolytic trials,10–12 and in patients with ST-segment elevation MI in the National Registry of Myocardial Infarction (NRMI) database from 1995 to 2004 (8.6%).7 More recently, however, the incidence of cardiogenic shock has fallen from about 8% to about 6% of MIs, with most of the change resulting from a decrease in cardiogenic shock developing after initial presentation, supporting the notion that early revascularization strategies are an important contributor to the decline.3 The most common cause of cardiogenic shock is left ventricular failure in the setting of an extensive acute MI, although a smaller infarction in a patient with previously compromised left ventricular function may also precipitate shock. Cardiogenic shock can also be caused by mechanical complications such as acute mitral regurgitation, rupture of the interventricular septum, or rupture of the free wall—or by large right ventricular infarctions. In a report of the SHOCK (SHould we emergently revascularize Occluded Coronaries for shocK) trial registry of 1160 patients with cardiogenic shock,2 78.5% of patients had predominant left ventricular failure, 6.9% had acute mitral regurgitation, 3.9% had ventricular septal rupture, 2.8% had isolated right ventricular shock, 1.4% had tamponade or cardiac rupture, and 6.5% had shock resulting from other causes. Other causes of cardiogenic shock include myocarditis, end-stage cardiomyopathy, myocardial contusion, septic shock with severe myocardial depression, myocardial dysfunction after prolonged cardiopulmonary bypass, valvular heart disease, and hypertrophic obstructive cardiomyopathy (Box 22.2). An important consideration is that some cardiogenic shock may have an iatrogenic component. Early diagnosis of impending shock or of patients at high risk for development of shock is essential, both to speed intervention and to avoid therapies that may worsen hemodynamics. In many cases of cardiogenic shock in the setting of MI, the diagnosis is not made until the patient has been triaged and admitted to an inpatient setting. Patients may have received early beta blockade or angiotensin-converting enzyme inhibition, therapies that may impact hemodynamics substantially. Patients may have cardiogenic shock at initial presentation, but most do not; shock usually evolves over several hours,15,16 suggesting that early treatment may potentially prevent shock. In fact, some data indicate that early thrombolytic therapy may decrease the incidence of cardiogenic shock.17 In the SHOCK trial registry, 75% of patients developed cardiogenic shock within 24 hours after presentation, with a median delay of 7 hours.2 Results from the GUSTO trial were similar;13 among patients with shock, 11% were in shock on arrival and 89% developed shock after admission. Risk factors for the development of cardiogenic shock in MI generally parallel those for left ventricular dysfunction and the severity of coronary artery disease. Shock is more likely to develop in patients who are elderly, are diabetic, and have anterior MI.5,15,18,19 Patients with cardiogenic shock are also more likely to have histories of previous infarction, peripheral vascular disease, and cerebrovascular disease.18,19 Decreased ejection fractions and larger infarctions (as evidenced by higher cardiac enzymes) are also predictors of the development of cardiogenic shock.18,19 Analysis from the GUSTO-3 trial has identified age, lower systolic blood pressure, heart rate, and Killip class as significant predictors of the risk for development of cardiogenic shock after presentation with acute MI.20 Use of a predictive scoring system derived from this study may be useful in identifying patients at high risk for the development of cardiogenic shock and targeting such patients for closer monitoring.20 Cardiogenic shock is most often associated with anterior MI. In the SHOCK trial registry, 55% of infarctions were anterior, 46% were inferior, 21% were posterior, and 50% were in multiple locations.2 These findings were consistent with those in other series.21 Angiographic evidence most often demonstrates multivessel coronary disease (left main occlusion in 20% of patients, three-vessel disease in 64%, two-vessel disease in 23%, and one-vessel disease in 13% of patients).22 The high prevalence of multivessel coronary artery disease is important because compensatory hyperkinesis normally develops in myocardial segments that are not involved in an acute MI, and this response helps maintain cardiac output. Failure to develop such a response, because of previous infarction or high-grade coronary stenoses, is an important risk factor for cardiogenic shock and death.16,23 Cardiac dysfunction in patients with cardiogenic shock is usually initiated by MI or ischemia. The myocardial dysfunction resulting from ischemia worsens that ischemia, creating a downward spiral24 (Fig. 22.1). When a critical mass of ischemic or necrotic left ventricular myocardium fails to pump, stroke volume and cardiac output decrease. Myocardial perfusion, which depends on the pressure gradient between the coronary arterial system and the left ventricle and on the duration of diastole, is compromised by hypotension and tachycardia, exacerbating ischemia. The increased ventricular diastolic pressures caused by pump failure reduce coronary perfusion pressure, and the additional wall stress elevates myocardial oxygen requirements, further worsening ischemia. Decreased cardiac output also compromises systemic perfusion. When myocardial function is depressed, several compensatory mechanisms are activated, including sympathetic stimulation to increase heart rate and contractility and renal fluid retention to increase preload. These compensatory mechanisms may become maladaptive and can actually worsen the situation when cardiogenic shock develops. Increased heart rate and contractility increase myocardial oxygen demand and exacerbate ischemia. Fluid retention and impaired diastolic filling caused by tachycardia and ischemia may result in pulmonary congestion and hypoxia. Vasoconstriction to maintain blood pressure increases myocardial afterload, further impairing cardiac performance and increasing myocardial oxygen demand. This increased demand, in the face of inadequate perfusion, worsens ischemia and begins a vicious cycle that will end in death if not interrupted (see Fig. 22.1). The interruption of this cycle of myocardial dysfunction and ischemia forms the basis for the therapeutic regimens for cardiogenic shock. Not all patients fit into this classic paradigm. In the SHOCK trial, the average systemic vascular resistance (SVR) was not elevated, and the range of values was wide, suggesting that compensatory vasoconstriction is not universal. Some patients had fever and elevated white blood cell counts along with decreased SVR, suggesting a systemic inflammatory response syndrome.25 This has led to an expansion of the paradigm to include the possibility of the contribution of inflammatory responses to vasodilation and myocardial stunning, leading clinically to persistence of shock (Fig. 22.2).25 Supporting this notion is the fact that the mean ejection fraction in the SHOCK trial was only moderately decreased (30%), suggesting that mechanisms other than pump failure were operative.25 Immune activation appears to be common to a number of different forms of shock. Activation of inducible nitric oxide synthase (iNOS) with production of nitric oxide and peroxynitrate has been proposed as one potential mechanism. Figure 22.2 Expansion of the pathophysiologic paradigm of cardiogenic shock to include the potential contribution of inflammatory mediators. Inhibition of nitric oxide, however, has not been shown to be beneficial in patients with cardiogenic shock.149 iNOS, inducible nitric oxide synthase; LVEDP, left ventricular end-diastolic pressure; NO, nitric oxide; ONOO−, peroxynitrite; SVR, systemic vascular resistance. (Adapted from Hochman JS: Cardiogenic shock complicating acute myocardial infarction: Expanding the paradigm. Circulation 2003;107:2999.) Cardiogenic shock is characterized by both systolic and diastolic myocardial dysfunction.16,26 Progressive myocardial necrosis has been observed consistently in clinical and pathologic studies of patients with cardiogenic shock.16,27 Patients who develop shock after admission often have evidence of infarct extension, which can result from reocclusion of a transiently patent infarct artery, propagation of intracoronary thrombus, or a combination of decreased coronary perfusion pressure and increased myocardial oxygen demand.18,19 Myocytes at the border zone of an infarction are more susceptible to additional ischemic episodes; therefore, these adjacent segments are at particular risk.28 Mechanical infarct expansion, which is seen most dramatically after extensive anterior MI, can also contribute to late development of cardiogenic shock.18,29 Ischemia remote from the infarct zone may be particularly important in producing systolic dysfunction in patients with cardiogenic shock.23,30 Patients with cardiogenic shock usually have multivessel coronary artery disease,2,16 with limited vasodilator reserve, impaired autoregulation, and consequent pressure-dependent coronary flow in several perfusion territories.31 Hypotension and metabolic derangements thus have the potential to impair the contractility of noninfarcted myocardium in patients with shock.32 This can limit hyperkinesis of uninvolved segments, a compensatory mechanism typically seen early after MI.23,30 Myocardial diastolic function is also impaired in patients with cardiogenic shock. Myocardial ischemia causes decreased compliance, increasing the left ventricular filling pressure at a given end-diastolic volume.33,34 Compensatory increases in left ventricular volumes to maintain stroke volume further increase filling pressures. Elevation of left ventricular pressures can lead to pulmonary edema and hypoxemia (see Fig. 22.1). Tissue hypoperfusion and consequent cellular hypoxia lead to anaerobic glycolysis, with depletion of adenosine triphosphate and intracellular energy reserves. Anaerobic glycolysis also causes accumulation of lactic acid and resultant intracellular acidosis. Failure of energy-dependent ion transport pumps decreases transmembrane potential, causing intracellular accumulation of sodium and calcium and myocyte swelling.35 Cellular ischemia and intracellular calcium accumulation can activate intracellular proteases.36 If the ischemia is severe and prolonged enough, myocardial cellular injury can become irreversible, with the classic pattern of myonecrosis: mitochondrial swelling, accumulation of denatured proteins and chromatin in the cytoplasm, lysosomal breakdown, and fracture of the mitochondria, nuclear envelope, and plasma membrane.35,36 Accumulating evidence indicates that apoptosis (programmed cell death) may also contribute to myocyte loss in MI.28,36,37 Although myonecrosis clearly outweighs apoptosis in the core of an infarcted area, evidence for apoptosis has been found consistently in the border zone of infarcts after ischemia and reperfusion and sporadically in areas remote from the ischemia area.28,37 Activation of inflammatory cascades, oxidative stress, and stretching of myocytes have been proposed as mechanisms that activate the apoptotic pathways.36,37 Although the magnitude of apoptotic cell loss in MI remains uncertain, inhibitors of apoptosis have been found to attenuate myocardial injury in animal models of postischemic reperfusion; these inhibitors may also have therapeutic potential for myocyte salvage after large infarctions.37 A key to understanding the pathophysiology and treatment of cardiogenic shock is to realize that large areas of nonfunctional but viable myocardium can also cause or contribute to the development of cardiogenic shock in patients after MI (Fig. 22.3). This reversible dysfunction can be described in two main categories: stunning and hibernation. Myocardial stunning represents postischemic dysfunction that persists despite restoration of normal blood flow; eventually, however, myocardial performance recovers completely.38 Originally defined in animal models of ischemia and reperfusion,39 stunning has been recognized in the clinical arena.38,40 Direct evidence for myocardial stunning in humans has been found using positron emission tomography (PET) scanning in patients with persistent wall motion abnormalities after angioplasty for acute coronary syndromes; perfusion measured by 13N-ammonia was normal in the presence of persistent contractile dysfunction.41 The pathogenesis of stunning has not been conclusively established but appears to involve a combination of oxidative stress,42 perturbation of calcium homeostasis, and decreased myofilament responsiveness to calcium.38,43,44 In addition to these direct effects, data from studies in isolated cardiac myocytes suggest that circulating myocardial depressant substances may contribute to contractile dysfunction in myocardial stunning.45 The intensity of stunning is determined primarily by the severity of the antecedent ischemic insult.38 Myocardial hibernation comprises segments with persistently impaired function at rest due to severely reduced coronary blood flow; inherent in the definition of hibernating myocardium is the notion that function can be normalized by improving blood flow.46–48 Hibernation can be seen as an adaptive response to reduce contractile function of hypoperfused myocardium and restore equilibrium between flow and function, thereby minimizing the potential for ischemia or necrosis.49 Revascularization of hibernating myocardium can lead to improved myocardial function,50 and improved function appears to translate into improved prognosis.51,52 Although hibernation is conceptually and pathophysiologically different from myocardial stunning, the two conditions are difficult to distinguish in the clinical setting and may in fact coexist.38,52 Repetitive episodes of myocardial stunning can coexist with or mimic myocardial hibernation.38,46,53 Consideration of myocardial stunning and hibernation is vital in patients with cardiogenic shock because of their therapeutic implications. Hibernating myocardium improves with revascularization, and stunned myocardium retains inotropic reserve and can respond to inotropic stimulation.38 In addition, the fact that the severity of the antecedent ischemic insult determines the intensity of stunning38 provides one of the rationales for reestablishment of patency of occluded coronary arteries in patients with cardiogenic shock. Finally, the notion that some myocardial tissue may recover function emphasizes the importance of measures to support hemodynamics and thus minimize myocardial necrosis in patients with shock. Cardiogenic shock is an emergency. The clinician must initiate therapy before shock irreversibly damages vital organs; at the same time, he or she must perform the clinical assessment required to understand the cause of shock and to target therapy to that cause. A practical approach is to make a rapid initial evaluation on the basis of a limited history, physical examination, and specific diagnostic procedures (Fig. 22.4).24 Cardiogenic shock is diagnosed after documentation of myocardial dysfunction and exclusion of alternative causes of hypotension such as hypovolemia, hemorrhage, sepsis, pulmonary embolism, tamponade, aortic dissection, and preexisting valvular disease. Echocardiography is an excellent initial tool for confirming the diagnosis of cardiogenic shock and ruling out other causes of shock (Box 22.3); therefore, early echocardiography should be routine. Echocardiography provides information on overall and regional systolic function, and can rapidly diagnose mechanical causes of shock such as papillary muscle rupture and acute mitral regurgitation, acute ventricular septal defect, and free wall rupture and tamponade.54,55 Unsuspected severe mitral regurgitation is not uncommon. In some cases, echocardiography may reveal findings compatible with right ventricular infarction. Invasive hemodynamic monitoring can be quite useful to exclude volume depletion, right ventricular infarction, and mechanical complications.16,35 The hemodynamic profile of cardiogenic shock includes a pulmonary capillary occlusion pressure greater than 15 mm Hg and a cardiac index less than 2.2 L/min/m2. It should be recognized that optimal filling pressures may be greater than 15 mm Hg in individual patients due to left ventricular diastolic dysfunction. Right-sided heart catheterization may reveal an oxygen step-up diagnostic of ventricular septal rupture or a large v wave that suggests severe mitral regurgitation. The hemodynamic profile of right ventricular infarction includes high right-sided filling pressures in the presence of normal or low occlusion pressures.56,57 Maintenance of adequate oxygenation and ventilation are critical; intubation and mechanical ventilation are often required, if only to reduce the work of breathing and facilitate sedation and stabilization before cardiac catheterization. Central venous and arterial access, bladder catheterization, and pulse oximetry are routine. Electrolyte abnormalities should be corrected. Hypokalemia and hypomagnesemia are predisposing factors to ventricular arrhythmias, and acidosis can decrease contractile function. Relief of pain and anxiety with morphine sulfate (or fentanyl if systolic pressure is compromised) can reduce excessive sympathetic activity and decrease oxygen demand, preload, and afterload. Arrhythmias and heart block may have major effects on cardiac output, and should be corrected promptly with antiarrhythmic drugs, cardioversion, or pacing. Cardiology consultation has been shown to be associated with improved outcomes in patients with MI and is strongly indicated in the setting of cardiogenic shock.58 In addition, measures proven to improve outcome after MI, such as nitrates, beta blockers, and angiotensin-converting enzyme inhibitors,59 have the potential to exacerbate hypotension in cardiogenic shock and should be stopped until the patient stabilizes. Following initial stabilization and restoration of adequate blood pressure, tissue perfusion should be assessed (see Fig. 22.4). If tissue perfusion remains inadequate, inotropic support or intra-aortic balloon pumping (IABP) should be initiated. If tissue perfusion is adequate but significant pulmonary congestion remains, diuretics may be employed. Vasodilators can be considered as well, depending on the blood pressure. The initial approach to the hypotensive patient should include fluid resuscitation unless frank pulmonary edema is present. Patients are commonly diaphoretic and relative hypovolemia may be present. In the original description of hemodynamic subsets in MI, approximately 20% of patients had low cardiac index and low PCWP; most had reduced stroke volume and compensatory tachycardia.60 Some of these patients would be expected to respond to fluid infusion with an increase in stroke volume, although the magnitude of such a response depends on the degree of ischemia and cardiac reserve.
Cardiogenic Shock
Definition
Epidemiology
Incidence
Etiology
Pathogenesis
Systemic Effects
Myocardial Pathology
Cellular Pathology
Reversible Myocardial Dysfunction
Clinical Assessment
Evaluation
Initial Management
Therapy
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Anesthesia Key
Fastest Anesthesia & Intensive Care & Emergency Medicine Insight Engine