206 Bleeding Disorders
 Key Points
Key Points• Bleeding disorders are common and can be classified as congenital or acquired.
• A thorough history, including medication history, can help screen for many potential bleeding disorders.
• The prothrombin time is a measurement of abnormalities in the tissue factor pathway, whereas the activated partial thromboplastic time is a measurement of abnormalities in the contact activation pathway.
• Hemophilia A and B are both X-linked conditions, and factor replacement may be needed as prophylaxis or treatment for bleeding. Recombinant factor VIIa can be used to treat patients with inhibitors.
• von Willebrand disease is the most common congenital bleeding disorder. Treatment options vary, depending on the severity of the condition and bleeding, but they include desmopressin, aminocaproic acid, tranexamic acid, and factor replacement.
• Common causes of acquired coagulopathies include trauma, massive transfusion, disseminated intravascular coagulation, drugs, and toxins.
• The definitive treatment of disseminated intravascular coagulation is reversal or treatment of the underlying cause.
Epidemiology
Under normal physiologic conditions, a constant balance exists between clot formation and clot breakdown, with a preference favoring anticoagulation.1 Hemostasis results from a complex interaction among the vascular endothelium, platelets, the coagulation cascade, and the fibrinolytic system. Any disorder that affects this interplay can alter the equilibrium, thereby producing either excessive thrombosis or excessive hemorrhage.
Broadly speaking, bleeding disorders can be either inherited or acquired. Of the inherited disorders, the von Willebrand syndromes and hemophilia A are the most common, with a prevalence between 1 per 100 individuals and 1 per 100,000 individuals. Less common inherited disorders include deficiencies of factors II, V, VII, X, XIII, and fibrinogen, with a prevalence between 1 and 2 per 100,000 individuals.2 The most common acquired bleeding disorders are drug induced, from therapeutic administration of agents expressly intended to decrease the risk of thrombosis. Oral anticoagulants are taken by up to 2% of the population and up to 8% of persons more than 65 years old, and the use of antiplatelet agents is vastly more common than that.3 Patients with inherited bleeding disorders will likely present to the emergency department (ED) for treatment of abnormal bleeding at some point during their lifetime,4 and ED visits for bleeding in patients who are taking antithrombotic agents are not uncommon.5 The clinician must be familiar with these disease states to provide prompt care because treatment delays can increase morbidity and mortality.4,6
Physiology and Biology of Hemostasis
Coagulation Cascade
The coagulation cascade (Fig. 206.1) is typically thought of as two pathways working separately. In reality, however, the tissue factor pathway (formerly called the extrinsic system) and the contact activation pathway (formerly called the intrinsic system) function intimately with each other to produce thrombin, a critical enzyme in the coagulation system.1,7
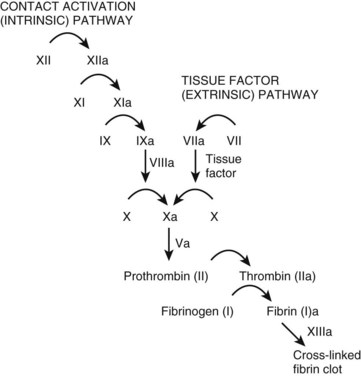
Tissue factor typically resides on smooth muscle cells, fibroblasts, and pericytes.8–10 Binding of tissue factor to factor VII results in an activated factor VII (factor VIIa). Factor VIIa subsequently catalyzes the conversion of factors IX and X to their active forms (factors IXa and Xa, respectively).
The contact activation pathway begins with the activation of factor XII by the high-molecular-weight enzymes kininogen and prekallikrein.1 This activation subsequently triggers a series of reactions leading to the conversion of X to Xa, the first step of the common pathway. The exact relevance of the contact activation pathway is debatable, however, because its activation is not needed in trauma-initiated coagulopathy.1 Furthermore, with the exception of a deficiency of factor XI, deficiencies in enzymes involved in the contact activation pathway generally do not produce substantial coagulopathies.1
Platelet Function
Following injury to the vascular endothelium, endothelin is released, resulting in vasoconstriction. In addition, injury to the vascular endothelium results in tissue factor exposure, with subsequent binding of platelets to the endothelium.8,10 Platelet adhesion primarily involves the binding of platelets to damaged endothelium by von Willebrand factor (vWF).11,12 Additional platelet activation occurs through the release of numerous mediators including adenosine diphosphate (ADP), epinephrine, thromboxane A2, and thrombin.11 Ultimately, platelet activation and aggregation require glycoprotein IIb/IIIa.11 Thrombus stability, however, requires fibrin, which is formed from the coagulation cascade on the surface of platelets.11
Differential Diagnosis and Medical Decision Making
The initial assessment of a patient with a known or suspected bleeding disorder begins with a thorough history and physical examination. Laboratory studies can be used to assess the severity of the disorder and to support a diagnosis. The initial laboratory studies that should be obtained when evaluating such patients include a complete blood count, prothrombin time (PT; a measure of the tissue factor pathway), and an activated partial thromboplastin time (aPTT; a measure of the contact activation pathway). A fibrinogen count can also assist in the initial management. These laboratory tests can help screen for clinically significant factor deficiencies, such as hemophilia A or B, as well as hypofibrinogenemic states. Additional tests may be indicated, depending on the condition of interest. For example, the concentration of D-dimers may be helpful in establishing the diagnosis of disseminated coagulation (DIC), whereas factor concentrations may be useful in assessing the severity of hemophilia. Patients with acute fulminant hepatic failure may demonstrate abnormalities similar to those seen in DIC. In such cases, factor V, VII, and VIII levels can be obtained. Although factor V levels are likely to be decreased in both DIC and liver failure, factor VIII levels are normal or elevated in liver failure–induced coagulopathy, yet markedly decreased in DIC-induced coagulopathy. Factor VII levels are also reduced in coagulopathy resulting from hepatic failure. Mixing studies can be performed to determine whether a factor deficiency or inhibitor is present in patients with prolonged aPTT.13 Examination of the peripheral blood film can be very helpful in patients with unexplained bleeding disorders.
Coagulation Cascade Defects
Platelet-related defects are discussed in Chapter 205.
Hereditary Defects
Hemophilia A and B
Hemophilia A and hemophilia B are X-linked recessive disorders involving factors VIII and IX, respectively. Clinically, hemophilia B is indistinguishable from hemophilia A. However, nearly half of all cases of hemophilia A, the most common form of hemophilia, represent de novo mutations, in which boys are born to parents without the disease.1 These disorders can be classified as mild, moderate, or severe, corresponding to a plasma coagulation factor concentration of 6% to 30%, 2% to 5%, or 1% or lower, respectively.14 Those patients with a mild form of the disease generally have bleeding only after trauma or surgery. In contrast, patients with the severe form of the disease have an average of 20 to 30 bleeding episodes annually, and bleeding occurs spontaneously or after minor trauma.14 Hemarthrosis is one of the most common forms of bleeding in severe forms of hemophilia.1 Historically, treatment involved transfusions of plasma concentrates of coagulation factors.14 However, such an approach was associated with the acquisition of numerous infectious diseases, including human immunodeficiency virus infection or hepatitis, as well as the development of inhibitors to the clotting factors.14,15 Now, both plasma and recombinant-derived factor VIII and IX exist. Treatment can be administered as an on-demand regimen, in which therapy is given based on bleeding, or in a prophylactic regimen. Because the plasma half-life of factor IX is longer than of factor VIII (18 to 24 hours versus 8 to 12 hours), dosing of factor replacement is less frequent for hemophilia B than for hemophilia A.16 Table 206.1 shows a common treatment regimen. Minor bleeding requires factor replacement to achieve factor concentrations of 30% of normal (typically 30 units/kg). In contrast, major hemorrhagic events, including hemarthrosis and large muscle bleeding, requires factor replacement to achieve factor concentrations of at least 50% (typically 50 units/kg). Life-threatening hemorrhage requires factor replacement to achieve factor concentrations of at least 80% (typically 80 units/kg).16 In general, each unit per kilogram of body weight of factor VIII increases plasma factor VIII concentrations by 2%, whereas each unit per kilogram of body weight of factor IX increases plasma factor IX concentrations by 1%.16 In patients with life-threatening bleeding, however, in vivo factor concentrations should be followed to ensure adequate replacement. When patients with hemophilia A have developed inhibitors to factor VIII, recombinant factor VIIa (rFVIIa) (90 mcg/kg) has been used.4
Table 206.1 Common Treatment Strategics for coagulopathies*
| TREATMENT OPTIONS | NOTES | |
|---|---|---|
| Disease-Induced Coagulopathy | ||
| Factor V deficiency | FFP | |
| Factor VIII deficiency | Recombinant factor VIIIa | 1 U/kg IV increases plasma factor VIII concentrations by 2%. A typical dose will be 40 U/kg for severe bleeding. |
| Recombinant factor VIIa can be used for those with inhibitors to factor VIII | 90 mcg/kg IV | |
| Factor IX deficiency | Recombinant factor IXa | 1 U/kg IV increases plasma factor IX concentrations by 1%. A typical dose will be 80 U/kg for severe bleeding. |
| Factor X deficiency | FFP or PCC | |
| Factor XI deficiency | FFP | |
| von Willebrand disease | DDAVP Aminocaproic acid Tranexamic acid | 0.3 mcg/kg IV 50-60 mg/kg IV q4-6h 10-15 mg/kg IV q8-12h |
| Congenital fibrinogen disorders | FFP Cryoprecipitate Fibrinogen concentrates† | |
| Coagulopathy of trauma | FFP Platelets | |
| Disseminated intravascular coagulation | Cryoprecipitate Platelets | Primary treatment is to treat the underlying cause |
| Transfusion-related coagulopathy | FFP Platelets | |
| Drug/toxin–Induced Coagulopathy | ||
| Heparin | Protamine | 1 Unit reverses 100 U UFH |
| Factor Xa inhibitors | rFVIIa FFP | 90 mcg/kg IV |
| Warfarin | Vitamin K, FFP, PCC | |
| Crotalid envenomation | Crotalidae polyvalent immune Fav (ovine); CroFab | |
DDAVP, Desmopressin; FFP, fresh frozen plasma; PCC, pool complex concentrates.
* Many etiologies of coagulopathy do not require factor replacement or transfusion. If these products are to be administered, the specific agent is listed in the table. However, for many conditions, no blood product should be transfused. The text can help guide when specific therapies should be administered.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


