Chapter 45 Asthma
The term status asthmaticus has been used to denote a more severe form of asthma attack, but its definition varies widely among different authors. To some authors, status asthmaticus is an asthma attack that does not respond to initial treatments with bronchodilators,1,2 whereas to others it indicates severe asthma that leads to respiratory failure and requires mechanical ventilatory support.3 For the purposes of this text, status asthmaticus is defined as an asthma attack that fails to respond to initial doses of nebulized β2-adrenergic and anticholinergic agents and systemic corticosteroid drugs and that requires admission to the hospital for continuation of treatment. Patients who experience relentless progression of respiratory signs and symptoms and require admission to the intensive care unit (ICU) are reported as having near-fatal asthma.4
Epidemiology and Risk Factors
Asthma is the most common chronic illness in childhood, affecting approximately 6.7 million children and adolescents in the United States, or 9.1% of persons younger than 18 years.5 Asthma is also a very common discharge diagnosis in children’s hospitals, accounting for approximately 5.6% of all hospital admissions and more than 150,000 admissions each year in the United States alone.5 The prevalence of asthma worldwide is highly variable, with greater than twentyfold differences in prevalence of symptoms encountered among centers located in various parts of the world.6 The highest prevalence rates for asthma are found in the United Kingdom, Australia, New Zealand, and the Republic of Ireland; very low prevalence rates occur in eastern Europe, the Indian subcontinent, and China.6 Race is a significant factor in determining prevalence and severity of asthma in children and young adults. African Americans are 4.1 times more likely to require treatment for asthma in the emergency department, two times more likely to be hospitalized for asthma, and 7.6 times more likely to die compared with white persons.5 Socioeconomic status also has been shown to negatively correlate with asthma prevalence, morbidity, and mortality in the United States.7
Asthma prevalence has increased steadily during the past 27 years in the United States.5 However, while the rate of nonurgent asthma-related outpatient visits continues to increase, emergency department visits and death rates have steadily declined during the past decade.5
The incidence of asthma-related respiratory failure requiring mechanical ventilation is difficult to determine because of variability in diagnostic criteria and reporting practices. Nonetheless, up to 36% of adult patients admitted to an inner-city medical ICU with near-fatal asthma require invasive mechanical ventilation.8 This figure appears to be significantly lower for children, considering that only 22 (10.2%) of 237 patients treated for near-fatal asthma underwent mechanical ventilation during a 2-year period in the pediatric ICU (PICU) of a tertiary children’s hospital (A.T. Rotta, unpublished data), and that only 14 (8.6%) of 163 patients required intubation in another study.9
The majority of patients with asthma who experience respiratory failure or arrest do so during the first stages of therapy or prior to arrival in the emergency department.9 Therefore early identification and close monitoring of patients at high risk for near-fatal asthma could be advantageous. High-risk patients often have a history of ICU admissions,10 mechanical ventilation,2,10 seizures or syncope during an attack,11 PaCO2 greater than 45 torr,2,10 attacks precipitated by food,10 or a history of rapidly progressive and sudden respiratory deterioration.1 These patients are likely to use more than two canisters of β-agonist metered-dose inhalers per month12 and often are poorly compliant or are receiving insufficient steroid therapy.13,14 Denial or failure to perceive the severity of an attack are factors frequently associated with near-fatal asthma.15,16 Although unquestionably some patients at risk for near-fatal asthma simply ignore early warning signs and do not seek adequate therapy, a subgroup of patients actually lacks normal perception of disease severity. Some patients with near-fatal asthma exhibit reduced chemosensitivity to hypoxia and blunted perception of dyspnea.17 Other patients have a decreased perceptual sensitivity of inspiratory muscle loads and display abnormal respiratory-related evoked potentials.18
Although many of these high-risk factors are commonly present in patients with near-fatal asthma, they fail to identify a significant number of cases. In one study, 33% of patients who died of asthma were judged to have a history of trivial or mild asthma, whereas 32% had never been admitted to the hospital with an asthma exacerbation.1 Some of these patients may in fact have what likely represents a distinct clinical entity known as sudden asphyxial asthma, a condition marked by acute onset of severe airway obstruction and hypoxia that rapidly leads to cardiorespiratory arrest in patients known to have only mild asthma or no asthma history at all.19,20
Pathophysiology
Asthma is primarily an inflammatory disease and, as such, it is marked by highly redundant pathways and complex interactions among inflammatory cells, mediators, and the airway epithelium21 (Figure 45-1). Functionally, asthma is characterized by variable airflow obstruction and airway hyperresponsiveness associated with airway inflammation. Pathologically, it is marked by mast cell degranulation, accumulation of eosinophils and CD4 lymphocytes, hypersecretion of mucus, thickening of the subepithelial collagen layer, and smooth muscle hypertrophy and hyperplasia.22
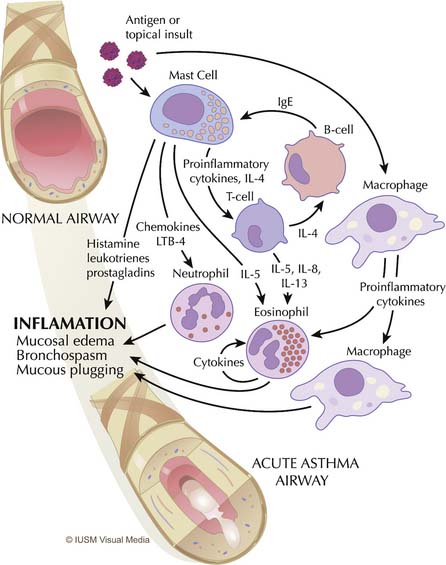
Mast cells, eosinophils, macrophages, and T lymphocytes are central to the derangements that occur during an acute attack (see Figure 45-1). The usual cascade begins with the activation and degranulation of mast cells in response to allergens or topical insults. The mast cells in turn promote activation of T lymphocytes by presenting these cells to the allergenic particles. The inflammatory process is then amplified by T-lymphocyte release of cytokines and chemokines. Increasing evidence exists that airway inflammation in asthma is the result of T-lymphocyte activation with the production of TH2 cytokines, such as interleukin (IL)-4, IL-5, IL-8, and IL-13.23 The presence of these TH2 cytokines leads to further augmentation of the inflammatory process through overexuberant production of immunoglobulin E (IgE) by B cells, stimulation of airway epithelial cells, and eosinophil chemotaxis. IgE stimulates mast cells to release leukotrienes, whereas interleukins (particularly IL-5) promote maturation and migration of activated eosinophils into the airway.24 This highly inflammatory milieu results in stimulation of airway epithelial cells and continued augmentation of the inflammatory process by further release of leukotrienes, prostaglandins, nitric oxide, adhesion molecules, and platelet-activating factor. This process results in overproduction of mucus and epithelial cell destruction that lead to airway plugging and denudation of the airway surface. Epithelial denudation is known to expose nerve endings, resulting in hyperirritable airways25 that become more susceptible to spasm and obstruction when challenged by subsequent exposure to allergens,26 inhaled irritants such as cigarette smoke and pollution,27 respiratory tract infections,28 psychological stress,29 and exercise,30 among other insults. Mucus itself is pathological in content,31,32 and mucus hypersecretion has been underappreciated as a cause of respiratory failure in persons with severe asthma, when in fact strong evidence exists that it may be a principal cause.31–33
Inflammation-mediated edema, mucus hypersecretion, airway plugging, and bronchospasm lead to the severe airway obstruction seen in patients with status asthmaticus and near-fatal asthma. The resulting obstruction and increased airway resistance create an impediment for inspiratory and expiratory gas flow, which leads to deranged pulmonary mechanics and increased lung volumes.32
Airway plugging can result in ventilation/perfusion mismatching and increased oxygen requirements. Hypoxemia is common in patients with a severe asthma attack, but it is generally easily corrected with supplemental oxygen34 and is only weakly correlated with pulmonary function abnormalities.35 More frequently, airway plugging and obstruction lead to regional alveolar hyperinflation associated with reduced perfusion, resulting in a significantly increased pulmonary dead space. Most patients with this condition exhibit an increased respiratory rate in attempt to achieve a higher minute volume and compensate for the ventilation abnormality. Unfortunately, in patients with more severe disease, airway obstruction also results in significant prolongation of expiratory time, which, coupled with initiation of inspiration prior to completion of the previous exhalation, leads to dynamic hyperinflation, gas trapping, and the development of abnormally high lung volumes36 (Figure 45-2).
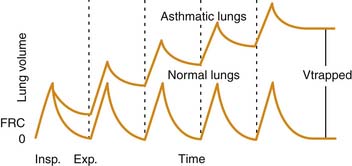
The higher lung volumes that result from incomplete alveolar emptying and dynamic hyperinflation serve as an adaptation mechanism to allow for higher expiratory flows than would have been possible at lower, more physiologic lung volumes. This higher expiratory flow is accomplished, however, at a high energy cost. Expiration becomes an active process, and the use of accessory muscles is required to overcome the high resistances to airflow both during inspiration and exhalation.37 During a severe attack, inspiratory transpulmonary pressures in excess of 50 cm H2O may be generated, compared with approximately 5 cm H2O during normal breathing.38 The increased muscle work is accompanied by an increase in blood flow to the diaphragm, but this flow often is insufficient to meet the much greater metabolic demands.39 Failure to promptly relieve the airway obstruction and reduce the work of breathing eventually leads to respiratory muscle fatigue, inadequate ventilation, and respiratory failure.
States of advanced airway obstruction and dynamic hyperinflation typical of severe asthma attacks have a significant impact on the circulatory system. The highly negative intrapleural pressures generated by spontaneously breathing patients during inspiration favor transcapillary edema fluid movement into the air spaces.37 They also cause a phasic increase in left ventricular afterload and a decrease in cardiac output40 that is clinically manifested as pulsus paradoxus.41 Right ventricular afterload may be increased during severe asthma as a result of pulmonary vasoconstriction related to hypoxia and acidosis. A state of increased pulmonary vascular resistance resulting from dynamic hyperinflation also can increase right ventricular afterload, further affecting cardiac output.41–43
Clinical Assessment
History
The child with an asthma exacerbation usually presents with complaints of difficulty breathing and shortness of breath. The presence of these complaints in a child known to have had previous asthma exacerbations is highly suggestive of the diagnosis. A significant percentage of children have a history of a coexisting viral upper respiratory infection, whereas some describe exposure to known allergic triggers. Circumstances permitting, time should be taken to inquire about the presence of high-risk factors (Box 45-1) for near-fatal asthma and the adequacy of maintenance intercrisis therapy.
Physical Examination
Wheezing, which is a common clinical finding in patients with acute asthma exacerbations, is the audible manifestation of the transmitted turbulence to airflow in the intrathoracic intrapulmonary airways. Wheezing may be predominantly expiratory as a result of the dynamic phasic compression of conducting airways, but it also can be biphasic. Wheezing in persons with severe asthma usually is symmetrical. An asymmetrical distribution suggests regional mucous plugging, atelectasis, pneumothorax, or the presence of a foreign body. The degree of wheezing correlates poorly with disease severity,44 because wheezes are heard only in the presence of airflow. As such, a patient with severe airway obstruction and very limited airflow may have a silent chest upon arrival at the emergency department, but loud wheezes may develop after effective therapy is instituted. Likewise, in a patient with loud wheezes that continue to worsen, a silent chest may develop as a prelude to respiratory failure.
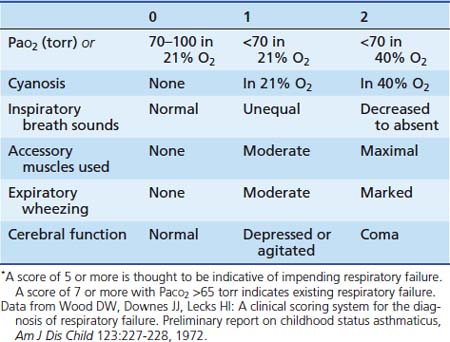
An objective assessment of disease severity is important in evaluating a patient’s response to therapy. Wood and colleagues45 developed a practical clinical asthma score composed of five variables with three different grades that allows for semiquantitative assessment of disease severity (Table 45-1). This clinical asthma score has been shown to correlate well with the need for prolonged bronchodilator therapy and hospitalization.46 However, although clinical asthma scores seem to be useful for assessing the severity of an attack, they are not as effective in prospectively identifying patients who require prolonged hospitalization or in whom complications and subsequent disability develop.47,48
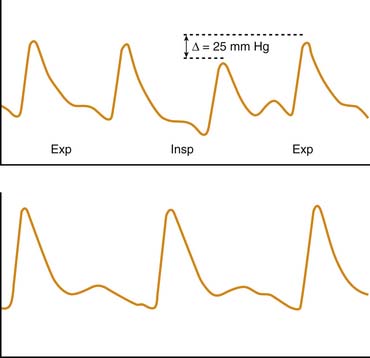
A less frequently used but more objective method of assessing disease severity and progression in patients with severe asthma is measurement of the pulsus paradoxus. Originally described by Adolf Kussmaul49 in a patient with constrictive pericarditis, pulsus paradoxus also is observed in conditions in which pleural pressure swings are exaggerated, such as status asthmaticus and near-fatal asthma. The simplest definition of pulsus paradoxus is an exaggeration of the physiologic inspiratory decrease in systolic blood pressure50 (Figure 45-3). It has been suggested that the term pulsus paradoxus is inappropriate to describe this phenomenon,51 because an accentuated inspiratory decrease in systolic pressure in the same direction as the normally occurring change cannot be described as a paradox. However, the true paradox described by Kussmaul49 was “the presence of a pulse slight and irregular, disappearing during inspiration and returning upon expiration despite the continued presence of the cardiac impulse during both respiratory phases.”50 Several mechanisms have been implicated in the occurrence of pulsus paradoxus in persons with asthma, and it is likely that various mechanisms contribute differently depending on the adequacy of intravascular volume, the magnitude of pleural pressure swings, the degree of pulmonary hyperinflation, and the state of cardiac contractility. These mechanisms include increased left ventricular afterload from highly negative intrapleural pressure52; decreased left ventricular preload as a result of inspiratory blood pooling in the pulmonary vasculature53; impaired left ventricular diastolic filling caused by a leftward shift of the interventricular septum resulting from increased venous return to the right heart54; constraint of cardiac filling because of longitudinal inspiratory deformation of the pericardium41; and increased right ventricular afterload with decreased filling of the left ventricle as a result of hyperinflation, acidosis, and hypoxia.55 The pulsus paradoxus can be measured easily in a patient who is spontaneously breathing by transducing pressure signals from an indwelling arterial catheter or by using a manual sphygmomanometer. In the latter technique, the cuff is inflated 20 mm Hg above the systolic pressure and then deflated until the first Korotkoff sounds are heard (systolic blood pressure). Initially, Korotkoff sounds are heard only during expiration. The cuff is then carefully deflated until the point where the sounds are heard equally during both inspiration and expiration. The difference between the highest systolic pressure and the pressure at which all Korotkoff sounds are heard is the magnitude of the pulsus paradoxus. During normal breathing, this difference is less than 5 mm Hg, but it is generally greater than 10 mm Hg during acute asthma exacerbations and greater than 20 mm Hg in patients with more severe disease.56 Changes in the magnitude of pulsus paradoxus during the course of therapy are good indicators of disease severity and clinical response to treatment.41,56
Laboratory Data
Arterial Blood Gas Analysis
Arterial blood gas measurements provide objective information on the adequacy of ventilation and oxygenation of the patient with asthma. The typical blood gas abnormality encountered in the early phase of asthma is relative hypoxemia with hypocapnia (PaCO2 <35 torr), reflecting hyperventilation.57 With worsening of airway obstruction, PaCO2 measurements return to the normal range of approximately 40 torr. However, this “normal” PaCO2 should not be viewed as reassuring when taken in the context of prolonged expiratory time, tachypnea, and accessory muscle use.58 In fact, PaCO2 greater than 40 torr in a patient with status asthmaticus should be interpreted as a sign of evolving respiratory muscle fatigue and warrants close clinical observation. Sicker patients often exhibit a mixed respiratory and metabolic acidosis.59 Lactic acidosis is frequently encountered in these patients and is thought to represent exaggerated lactate production by the respiratory muscles and tissue hypoxia.60
Muscle Enzymes
At least one third of patients with acute severe asthma exhibit an elevated plasma creatine kinase (CK) level.61 Although such elevations seem to be more pronounced in patients with marked acidemia or in those presenting with more severe respiratory insufficiency, a convincing correlation between disease severity and CK elevation has not been established.61 Myoglobin, a heme protein present in skeletal and cardiac muscle, is often also elevated in patients with near–fatal asthma.61 Elevations of CK–myocardial bound (CK–MB) isoenzyme develop in some patients with near–fatal asthma, suggesting a possible myocardial injury. This scenario certainly is plausible, considering that many patients are hypoxemic, acidotic, have high myocardial energy demand, and are receiving medications with adverse cardiac effects. However, plasma myoglobin and CK–MB elevations cannot be solely attributed to myocardial injury because the lungs and respiratory muscles are also known sources of these substances.62 Cardiospecific troponin T is a very sensitive and specific marker of myocardial cell damage63 and should be used preferentially to address the question of cardiomyocyte involvement in patients with severe asthma.
Electrocardiography
Patients with status asthmaticus or near-fatal asthma with significant airway obstruction and hyperinflation may exhibit a change in the mean frontal P–wave vector. A P–wave axis greater than 60 degrees has been associated with hyperinflation in both pediatric and adult patients with airway obstruction and is thought to represent positional atrial changes caused by inferior displacement of the diaphragm.64
Twelve–lead electrocardiography and continuous cardiac monitoring are valuable tools in the care of patients with near-fatal asthma in the ICU environment. These patients usually receive high doses of β-agonist drugs and may show evidence of hypokalemia (low–voltage T waves) or cardiac arrhythmias.65,66 The already increased myocardial energy demand resulting from airway obstruction is compounded by the chronotropic and vasodilatory effects inherent to β–agonist drugs and may lead to myocardial ischemia, particularly in adult patients with restricted coronary perfusion. Pediatric patients also may exhibit electrocardiographic (ECG) and enzymatic evidence of myocardial ischemia, particularly during treatment with intravenously administered isoproterenol.67 However, despite the fact that a study reported that a high percentage (66%) of patients exhibited nonspecific ST segment changes or other criteria suggestive of ischemia, these changes were not well correlated with initiation of terbutaline therapy or elevations in cardiac troponin T.68
Treatment
Initial Management in the Emergency Department
Patients with moderate or severe acute asthma also should receive a dose of systemic corticosteroid in the emergency department, which usually is administered prior to the second dose of albuterol. Prednisone (2 mg/kg) can be administered orally and is generally well tolerated. Oral prednisone is superior to inhaled fluticasone in children with severe asthma as evidenced by greater improvement in pulmonary function and lower hospitalization rates.69 The role of corticosteroid drugs in reversing an acute asthma attack in the emergency department has been the subject of debate, considering that these drugs require at least 4 to 6 hours for peak effects to be manifested.70 However, regardless of considerations about onset of action, acute suppression of inflammation is a cornerstone of acute asthma treatment and should be initiated as early as possible. Sicker patients with severe asthma exacerbations, those unable to tolerate oral medication because of respiratory distress or emesis, or those with a history of nausea during intensive β–agonist therapy should be given parenteral corticosteroid drugs such as methylprednisolone (2 mg/kg administered intravenously, followed by 0.5 to 1 mg/kg/dose administered intravenously every 6 hours).
Inhaled or nebulized anticholinergic agents such as ipratropium bromide are now considered an important adjunct in the treatment of persons with moderate and severe asthma exacerbations in the emergency department. In patients treated with one dose of a corticosteroid, use of ipratropium bromide (500 μg/2.5 mL) in conjunction with the second and third albuterol (salbutamol) doses has been associated with greater clinical improvement71 and reduced hospitalization rates compared with corticosteroid and albuterol (salbutamol) alone.72
Admission Criteria
The majority of patients with an acute asthma exacerbation respond to treatment in the emergency department and are discharged home. Among patients whose symptoms persist despite initial treatment, most can be safely managed in the general pediatric inpatient ward. Indications for hospitalization after treatment in the emergency department are loosely defined but may include (1) an inadequate response to three or four aerosol treatments; (2) relapse within 1 hour of receiving treatment with aerosols and steroids; (3) persistent SpO2 measurements of less than 91% in room air; (4) the need for oxygen therapy; (5) a significant reduction in peak expiratory flow rate; (6) having unreliable family support or being unable to comply with outpatient treatment; and (7) multiple visits for the same episode.73,74 Patients who require higher levels of monitoring or more invasive and aggressive treatment or who deteriorate during hospitalization in the general pediatric ward should be admitted to the PICU.
Management in the Intensive Care Unit
Oxygen
Sick patients with asthma are likely to exhibit hypoxemia as a result of intrapulmonary shunts caused by mucus plugging and atelectasis. Treatment with β–agonist agents also can contribute to hypoxemia by abolishing regional pulmonary hypoxic vasoconstriction and increasing intrapulmonary shunt.75,76 Therefore humidified oxygen should be offered both as a carrier gas for nebulizations and continuously between treatments.77 Supplemental oxygen can be safely incorporated into the treatment algorithm because, unlike in some adult patients with severe chronic obstructive pulmonary disease78 or asthma,79 no evidence exists to suggest that supplemental oxygen suppresses the respiratory drive in children with near–fatal asthma.
Corticosteroids
Corticosteroid drugs play a central role in the treatment of patients with status asthmaticus and near-fatal asthma, considering that these conditions are predominantly inflammatory in nature. Glucocorticosteroid agents modulate airway inflammation by a number of mechanisms, including direct interaction with cytosolic receptors and glucocorticosteroid response elements in gene promoters and indirect effects on binding of transcription factors, such as nuclear factor–κB, and on other cell signaling processes, such as posttranscriptional events.80 Gene products suppressed by glucocorticosteroid agents include a wide range of cytokines (IL–1, IL–2, IL–3, IL–4, IL–5, IL–6, IL–7, IL–8, IL–11, IL–12, IL–13, tumor necrosis factor–α, and granulocyte-macrophage colony–stimulating factor), adhesion molecules (intracellular adhesion molecule–1 and vascular cell adhesion molecule-1), and inducible enzymes, including NO synthase and cyclooxygenase–2.81 Transcription of other genes, such as lipocortin–1 and the β2–adrenergic receptor, may be enhanced.81 Glucocorticosteroid agents also decrease airway mucus production, reduce inflammatory cell infiltration and activation, and attenuate capillary permeability.82–85
In children with status asthmaticus or near–fatal asthma, glucocorticosteroid drugs should be administered by the IV route. The oral route can be used in selected cases, but inhaled glucocorticosteroid drugs play no role in the treatment of the sick hospitalized patient.21,69 The most common agent used in the United States is methylprednisolone because of its wide availability as an IV preparation and lack of mineralocorticoid effects. The usual dose of methylprednisolone is 0.5 to 1 mg/kg/dose, administered intravenously every 6 hours. Hydrocortisone, an agent with both glucocorticoid and mineralocorticoid activity, can be used as an alternative at doses of 2 to 4 mg/kg/dose, administered intravenously every 6 hours. Short courses of steroids usually are well tolerated without significant adverse effects.84 However, hypertension, hyperglycemia, mood disorders, and serious viral infections, such as fatal varicella, have been reported in previously well patients with asthma who have received glucocorticosteroid drugs.84,86,87 Duration of corticosteroid therapy is dictated by the severity of illness and the clinical response. Once initiated, treatment in patients with status asthmaticus or near-fatal asthma is generally continued for 5 to 7 days. Longer treatment courses necessitate gradual weaning of the drug to decrease the chances of symptomatic adrenal insufficiency or relapse. Prophylaxis with an H2 blocker should be considered because of the possibility of steroid-associated gastritis and gastric perforation.88
β–Agonists
The β–agonist properties of the sympathomimetic agents cause bronchial smooth muscle relaxation and hence bronchodilatation. These agents also can increase diaphragmatic contractility, enhance mucociliary clearance, and inhibit bronchospastic mediators from mast cells.89 Therefore β–agonists, along with systemic corticosteroids, are the mainstay of pharmacotherapy in persons with near-fatal asthma. β2–receptor selectivity is desirable to avoid adverse effects of nonselective α– and β1–adrenergic receptor stimulation. However, despite β2 selectivity, cardiovascular adverse effects remain a dose–limiting factor. The relative potency of various agents for the β2 receptor is as follows: isoproterenol > fenoterol > albuterol > terbutaline > isoetharine > metaproterenol.90 Of these agents, only albuterol and terbutaline are widely used in clinical practice, with some centers still using isoproterenol in selected occasions.
Once bound to the β–adrenergic receptor, β–agonists activate adenyl cyclase, resulting in increased intracellular cyclic adenosine monophosphate (cAMP) levels, which leads to bronchial and vascular smooth muscle relaxation. Dose-response curves demonstrate that large dose increases fail to enhance bronchodilation significantly; however, as the degree of bronchial constriction increases, the bronchodilation dose–response curve shifts to the right, indicating the need for a higher dose to achieve the desired response.90
The most frequent untoward adverse effects of β–agonist agents are skeletal muscle tremor, nausea, and tachycardia. These adverse effects are common to both nonselective and selective β–agonist drugs administered by IV and inhalational routes. Other cardiovascular adverse effects include blood pressure instability (predominantly diastolic hypotension) and cardiac dysrhythmias.91,92 Myocardial ischemia has been well documented as a complication of IV isoproterenol administration to children with near-fatal asthma.67,93 However, continuous IV infusions of terbutaline appear to be safe and are not associated with significant cardiotoxicity.68 Prolongations of the QTc interval and hypokalemia have been observed during IV infusions of β–agonist drugs.94 Hypokalemia occurs in the setting of relatively stable total body potassium and is the result of intracellular potassium shifting that results, at least in part, from an increased number of sodium-potassium pumps and not from augmented potassium elimination.95 Therefore supraphysiologic potassium supplementation is rarely necessary. A less frequently recognized adverse effect of β–agonist agents is hypoxemia, which is likely related to decreased regional hypoxic pulmonary vasoconstriction in areas of atelectasis and the resultant increased intrapulmonary shunt.75,76
Albuterol (Salbutamol)
Albuterol is the most β2–specific aerosol agent available in the United States. It usually is administered every 20 minutes during the initial phase of treatment at a dose of 0.05 to 0.15 mg/kg. The optimal dose and frequency of albuterol are controversial because less than 1% of the nebulized drug is deposited in the lung.96 Moreover, spontaneous tidal volume, breathing pattern, and technique are unpredictable yet major determinants of drug delivery. After the initial series of three albuterol treatments, continuous albuterol nebulization should be started for patients who require nebulization treatments more frequently than every 1 hour.
Continuous albuterol nebulization appears to be superior to repeated intermittent dosing and has not been shown to cause significant cardiotoxicity.97–99 A small prospective randomized study in children with near–fatal asthma and impending respiratory failure indicated that children treated with continuous albuterol nebulization had more rapid clinical improvement and shorter hospitalizations compared with children treated with intermittent albuterol doses.98 Continuous administration of albuterol also was associated with more efficient allocation of respiratory therapists’ time98 and could offer the added advantage of more hours of uninterrupted sleep to patients who often are already exhausted.100 The usual dose of continuously administered albuterol ranges between 0.15 and 0.45 mg/kg/h, with a maximum dose of 20 mg/h. Higher doses of albuterol have been used in patients who are unresponsive to standard treatment.51 However, we do not support this practice, because the intensification of adverse effects can outweigh any small incremental gain in bronchodilatation.
The availability of levalbuterol has generated some controversy. Albuterol is a 50:50 mixture of R–albuterol (levalbuterol), the active enantiomer that causes bronchodilation, and S–albuterol, which was thought to be inactive in humans. The U.S. Food and Drug Administration has approved levalbuterol, the pure R–isomer, as a preservative–free nebulizer solution.101 The purported advantage of using levalbuterol over albuterol stems from the fact that S–albuterol may not be completely inert and has a longer elimination half–life than R–albuterol.102,103 However, the notion that S–albuterol is not inert and that it is capable of clinically significant adverse effects is not universally accepted.104–106 A large randomized controlled trial of levalbuterol versus racemic albuterol in children with asthma demonstrated a decreased rate of hospitalization in patients treated with levalbuterol. However, this study had methodological problems, as the primary outcome variable (rate of hospital admission) was left to the discretion of the treating physicians and none of the secondary outcome variables were significantly different between treatment groups once the patients had been admitted to the hospital.107 More recent randomized clinical studies in children with asthma failed to show definitive evidence that levalbuterol is superior to a regular racemic albuterol.108,109 Furthermore, although the cost of levalbuterol has decreased significantly in the past few years, this drug continues to be more expensive than albuterol (C. A. Thomas, PharmD, Riley Hospital for Children, personal communication, 2011). Considering the lower cost of albuterol and the paucity of clinical evidence supporting the superiority of levalbuterol, we continue to favor albuterol as the routine bronchodilator of choice in children with near–fatal asthma.
Intravenously administered albuterol is not available in the United States. However, the efficacy of albuterol infusions in patients with severe asthma has been well established in countries where the IV preparation is available.110–112





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


