97 Ascites
 Definition and Diagnosis
Definition and Diagnosis
Ascites is the abnormal accumulation of fluid in the peritoneal cavity.1 Patients with ascites generally present on clinical examination with abdominal distention and a fluid wave or shifting dullness on abdominal percussion, but the abdominal examination findings also may be normal if the amount of ascites is not massive.
Diagnostic imaging can confirm the diagnosis of ascites. Ultrasonography is the easiest and most sensitive technique for the detection of ascitic fluid, being capable of visualizing very small volumes (5-10 mL). Computed tomography (CT) is also very sensitive for detecting ascites (Figure 97-1). Small amounts of ascitic fluid localize in the perihepatic area and in Morrison’s pouch (the hepatorenal space).
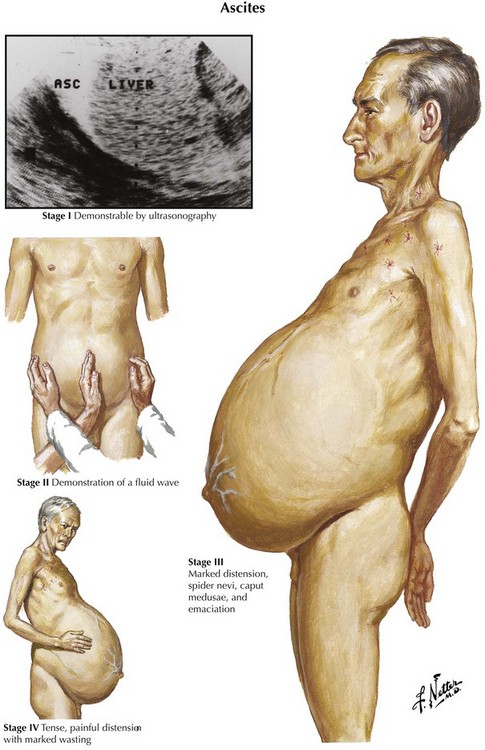
A diagnostic paracentesis (20 mL)2 is performed to determine the etiology of the ascites as well as to exclude or establish a diagnosis of spontaneous bacterial peritonitis (SBP). A diagnostic paracentesis should be performed in any person with new-onset ascites. Paracentesis to evaluate for SBP is also indicated for cirrhotic patients with known ascites who require hospitalization or sustain clinical deterioration, such as worsening encephalopathy or unexplained fever. A missed or delayed diagnosis of SBP can lead to sepsis and significant morbidity and mortality.
Peritoneal fluid from patients with new-onset ascites of unknown origin should be assayed for cell count, albumin level, culture, total protein concentration, Gram stain, and cytologic analysis.3 Serum albumin concentration should be measured as well.
The serum ascites albumin gradient (SAAG, serum albumin concentration—ascitic fluid albumin concentration) is the best diagnostic measure for classification of ascites (Table 97-1).4 The SAAG is very specific and sensitive for distinguishing ascites due to portal hypertension (SAAG > 1.1 g/dL) from that occurring as a result of other pathogenetic mechanisms such as inflammation or peritoneal malignancy (SAAG ≤ 1.1 g/dL). Ideally, specimens should be obtained simultaneously. In the past, ascites was classified as being an exudate (protein concentration ≥ 2.5 g/dL) or a transudate (protein concentration < 2.5 g/dL), but this classification scheme is no longer used because of its poor sensitivity and specificity.5 The total protein level may provide additional clues about diagnosis when used with the SAAG; that is, high SAAG and high protein concentration is seen in most cases of ascites due to hepatic congestion, whereas low serum ascites albumin gradient and high protein concentration characterizes malignant ascites. The terms high albumin gradient and low albumin gradient should replace the terms transudate and exudate in the description of ascites.
TABLE 97-1 Causes of Ascites Based on Normal or Diseased Peritoneum and Serum-to-Ascites Albumin Gradient (SAAG)
| Normal Peritoneum | |
| Portal Hypertension (SAAG > 1.1 g/dL) | Hypoalbuminemia (SAAG < 1.1 g/dL) |
| Hepatic congestion | Nephrotic syndrome |
| Congestive heart failure | Protein-losing enteropathy |
| Constrictive pericarditis | Severe malnutrition with anasarca |
| Tricuspid insufficiency | |
| Budd-Chiari syndrome | |
| Liver disease | Miscellaneous Conditions (SAAG < 1.1 g/dL) |
| Cirrhosis | Chylous ascites |
| Alcoholic hepatitis | Pancreatic ascites |
| Fulminant hepatic failure | Bile ascites |
| Massive hepatic metastases | Nephrogenic ascites |
| Urine ascites | |
| Ovarian disease | |
| Diseased Peritoneum (SAAG < 1.1 g/dL) | |
| Infections | Other Rare Conditions |
| Bacterial peritonitis | Familial Mediterranean fever |
| Tuberculous peritonitis | Vasculitis |
| Fungal peritonitis | Granulomatous peritonitis |
| HIV-associated peritonitis | Eosinophilic peritonitis |
| Malignant Conditions | |
| Peritoneal carcinomatosis | |
| Primary mesothelioma | |
| Pseudomyxoma peritonei | |
| Hepatocellular carcinoma | |
 Pathophysiology
Pathophysiology
Ascites is the most common complication related to liver disease and cirrhosis.6 It is associated with profound changes in the splanchnic and systemic circulation and with renal abnormalities (Figure 97-2). However, the pathogenesis of renal sodium retention and ascites formation in cirrhosis remains a subject of much controversy.
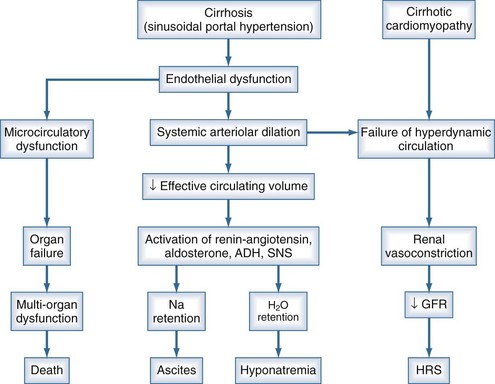
One accepted theory of ascites formation is the forward theory, which states that the development of ascites is related to the existence of severe sinusoidal portal hypertension that causes marked splanchnic arterial vasodilation and a forward increase in the splanchnic production of lymph.7 Splanchnic arterial vasodilation also produces arterial vascular underfilling, a significant reduction of the effective blood volume, and arterial hypotension. These pathophysiologic changes lead to compensatory activation of sodium- and water-retaining mechanisms (the renin-angiotensin-aldosterone system, sympathetic nervous system, and nonosmotic release of vasopressin) and promote ascites formation. Therefore, according to this theory, derangements in the splanchnic arterial circulation rather than the venous portal system are primary in the pathogenesis of ascites formation.8
Patients with advanced cirrhosis and portal hypertension often show an abnormal regulation of extracellular fluid volume, resulting in the accumulation of fluid as ascites, pleural effusion, or edema. The mechanisms responsible for ascites formation include alterations in the splanchnic circulation as well as renal functional abnormalities that favor sodium and water retention.9 The renal functional abnormalities occur in the setting of a hyperdynamic circulatory state that is characterized by increased cardiac output, decreased systemic vascular resistance, and activation of neurohormonal vasoactive systems. This circulatory dysfunction, due mainly to intense arterial vasodilation in the splanchnic circulation, is considered to be a primary feature in the pathogenesis of ascites.
The peripheral arterial vasodilation hypothesis incriminates relative underfilling of the arterial vascular compartment as the primary problem. Relative arterial underfilling leads to the same neurohumoral responses that occur in states characterized by low cardiac output (e.g., chronic congestive heart failure).10 Activation of the renin-angiotensin-aldosterone axis and the sympathetic system, as well as nonosmotic release of vasopressin, are well documented in cases of cirrhosis. This sequence of events results in renal water and sodium retention, failure to escape from the sodium-retaining effect of aldosterone, and renal resistance to atrial natriuretic peptide. Dilutional hyponatremia is the strongest predictor of the occurrence of hepatorenal syndrome.
The pathogenesis of peripheral arterial vasodilation in cirrhosis is not completely elucidated, but there is evidence for a major role of NO.11 Increased vascular NO production has been demonstrated in cirrhosis. In patients with ascites, the hepatic artery produces more NO than it does in patients without ascites. In a rat model of cirrhosis, normalization of vascular NO production with administration of a NO synthase inhibitor corrects the hyperdynamic circulation, improves sodium and water excretion, and decreases neurohumoral activation. This insight into the mechanisms of the peripheral arterial vasodilation in cirrhosis should provide new tools in the treatment of edema and ascites, a major cause of morbidity and mortality in patients with cirrhosis.
The generally accepted peripheral arterial vasodilation hypothesis seems to best explain the mechanism of sodium retention and other clinical findings such as hyperdynamic circulation in patients with cirrhosis. However, recent data in patients with pre-ascites or early ascites do not seem to conform to the peripheral arterial vasodilation hypothesis.12 Renal sodium handling abnormalities can be demonstrated in patients with cirrhosis prior to the development of ascites when these individuals are challenged with a sodium load. These changes are apparent even in the absence of systemic vasodilation or arterial underfilling. Therefore, an alternative hypothesis with a direct hepatorenal interaction, acting via sinusoidal portal hypertension and/or hepatic dysfunction as the effector mechanism, is proposed to be the initiating event promoting renal sodium retention in patients with cirrhosis. The second and later process is the development of systemic arterial vasodilation, possibly due to the presence of excess systemic vasodilators and/or decreased responsiveness of the vasculature to endogenous vasoconstrictors. These changes in turn lead to a relatively underfilled circulation with consequent activation of neurohumoral systems, promoting further renal sodium retention as described by the peripheral arterial vasodilation hypothesis. When compensatory natriuretic mechanisms fail, refractory ascites develops and hepatorenal syndrome sets in. Thus renal sodium retention in patients with cirrhosis is the result of an interplay of many factors; direct hepatorenal interaction predominates in the earlier stages of the cirrhotic process, whereas systemic vasodilation becomes a more important pathogenetic mechanism as the disease progresses.
 Etiology
Etiology
Most cases of ascites are due to liver disease. However, a number of disorders may be associated with ascites, and these include portal vein thrombosis, cardiac disorders (constrictive pericarditis, congestive heart failure), liver cancer, nephrotic syndrome, protein-losing enteropathy, and pancreatitis (see Table 97-1). Nonhepatic causes include cardiac failure, malignancy, renal failure, and intraabdominal inflammation. It is important to diagnose nonhepatic causes of ascites such as malignancy, tuberculosis, and pancreatic ascites, since these occur with increased frequency in patients with liver disease.
 Management
Management
Ascites is the most common presentation of decompensated cirrhosis. It occurs in more than half of all patients with cirrhosis, and its development heralds a poor prognosis (50% 2-year survival rate). Ascites is characterized by three grades of severity, and treatment is based on grade (Table 97-2). Effective first-line medical therapy for ascites includes dietary sodium restriction (2 g/d) and use of diuretics.13
TABLE 97-2 Grades of Ascites and Recommended Treatment
| Grade | Definition | Treatment |
|---|---|---|
| Grade 1 | Mild ascites only detectable by ultrasonographic examination | No specific treatment Dietary sodium restriction Careful follow-up |
| Grade 2 | Moderate ascites manifest by moderate symmetrical distention of the abdomen | Dietary sodium restriction Diuretics (spironolactone with or without furosemide, amiloride for patients with nonactivated renin-angiotensin-aldosterone system) |
| Grade 3 | Large or gross ascites with marked abdominal distention | Paracentesis (total or large-volume, with colloid volume expansion) Dietary sodium restriction Diuretics |
Adapted from Moore KP, Wong F, Gines P, et al. The management of ascites in cirrhosis: report on the Consensus Conference of the International Ascites Club. Hepatology 2003;38:258-66.
Medical Management
Management of uncomplicated ascites includes salt restriction, diuretics, and large-volume paracentesis (LVP) (Table 97-3). Diuretics are the mainstay of medical therapy in the treatment of ascites. Initially, an aldosterone antagonist (spironolactone) is used. Spironolactone competes with aldosterone for receptor sites in the distal renal tubules, increasing salt and water excretion and promoting retention of potassium and hydrogen ions. Spironolactone is usually initiated at a dose of 100 mg per day. The addition of a loop diuretic (e.g., furosemide) may be necessary in some cases to increase the natriuretic effect. The dosage of both the aldosterone antagonist and the loop diuretic should be increased sequentially until an adequate diuretic response is observed. Sodium restriction and diuretic therapy are initially effective in approximately 95% of patients. Water restriction is used only if persistent hyponatremia is present.
TABLE 97-3 Management of Uncomplicated Ascites
| General Management | Treat ascites once complications have been treated. | |
| Avoid NSAIDs. | ||
| Norfloxacin prophylaxis (400 mg PO once daily) in patients with an ascites protein level of <1.5 g/dL, impaired renal function (serum creatinine level ≥ 1.2 mg/dL, BUN ≥ 25 mg/dL, serum sodium level ≤ 130 mEq/L, or severe liver failure (CTP score ≥ 9 points with serum bilirubin level ≥ 3 mg/dL) | ||
| Specific Management | Salt restriction | 1-2 g/day Liberalize if restriction results in poor food intake. |
| Diuretics | Spironolactone based: spironolactone alone (start at 50-100 mg once daily, single morning dose) or: Spironolactone (50-100 mg once daily) + furosemide (start 20-40 mg once daily, single morning dose) | |
| LVP | Use as initial therapy only in patients with tense ascites; administer intravenous albumin (6-8 g/L of ascites removed). | |
| Follow-up and Goals | Adjustment of diuretic dosage should be performed every 4-7 days. | |
| Patient should be weighed at least weekly, and BUN, creatinine, and electrolytes measured every 1-2 weeks while adjusting dosage. | ||
| Double dosage of diuretics if: Weight loss < 4 lb (2 kg) a week and BUN, creatinine, and electrolytes stable | ||
| Halve the dosage of diuretics or discontinue if: Weight loss ≥ 1 lb (0.5 kg/day) or if there are abnormalities in BUN, creatinine, or electrolytes | ||
| Maximum diuretic dosage is spironolactone, 400 mg once daily, and furosemide, 160 mg once daily. | ||
BUN, blood urea nitrogen; CTP, Child-Turcotte-Pugh; LVP, large volume paracentesis; NSAIDs, nonsteroidal anti-inflammatory drugs; PO, orally.
Data from Garcia-Tsao G, Lim JK; Members of the Veterans Affairs Hepatitis C Resource Center Program. Management and treatment of patients with cirrhosis and portal hypertension: recommendations from the Department of Veterans Affairs Hepatitis C Resource Center Program and the National Hepatitis C Program. Am J Gastroenterol 2009;104:1802-29.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


