124 Agents with Primary Activity Against Gram-Positive Bacteria
The causes of nosocomial infections have changed in recent years. A 25-year study of nosocomial bacteremia demonstrated a change from Staphylococcus aureus and gram-negative bacilli as the predominant pathogens during the 1970s and 1980s to coagulase-negative staphylococci and Enterococcus, along with S. aureus and Pseudomonas aeruginosa, as the most common contemporary pathogens.1 The EPIC II study in 2007 demonstrated gram-positive organisms were associated with 47% of infections in the ICU.2 There can also be differences in the predominance of pathogens in different ICUs and different types of nosocomial infections. Nosocomial bacteremias are caused most often by coagulase-negative staphylococci and S. aureus in the medical ICU.3 S. aureus is the most common pathogen associated with nosocomial pneumonia and the fourth most common cause of skin and soft-tissue infections.3 Along with the increase in prevalence of gram-positive cocci in the ICU, staphylococci are becoming multidrug resistant. This chapter addresses gram-positive organisms and resistance issues associated with each of the antimicrobials with activity against these pathogens.
 Vancomycin
Vancomycin
Vancomycin was discovered in 1956 and marketed in 1958. Early preparations of the drug contained pyrogens and impurities that produced a brownish, muddy appearance that provided vancomycin’s nickname, “Mississippi mud.” In addition, these pyrogens and impurities caused high fevers, hypotension, severe phlebitis, and possibly nephrotoxicity.4
Mechanisms of Action And Resistance
Vancomycin inhibits synthesis of the cell wall by binding to the D-alanyl-D-alanine terminus of cell wall precursor units. Vancomycin is slowly bactericidal against dividing organisms except for Enterococcus and tolerant staphylococci, against which it is bacteriostatic.5 In 2006 the Clinical and Laboratory Standards Institute (CLSI) changed the vancomycin breakpoints against Staphylococcus aureus from ≤4 µg/mL to ≤2 µg/mL for susceptible strains. Intermediate susceptibility is now 4 to 8 µg/mL, and resistance to vancomycin is ≥16 µg/mL.6 The U.S. Food and Drug Administration (FDA) adopted these new breakpoints in 2008. The European Committee on Antimicrobial Susceptibility Testing (EUCAST) changed their vancomycin interpretations against S. aureus to ≤2 µg/mL as susceptible and >2 µg/mL as resistant. These changes in breakpoints will alter how literature is interpreted with respect to the frequency or prevalence of vancomycin-intermediate or vancomycin-resistant S. aureus over the past 30 years.
Five types of resistance for vancomycin have been isolated from enterococci: VanA, VanB, VanC, VanD, and VanE. The VanA phenotype confers high-level resistance to both teicoplanin (minimum inhibitory concentrations [MICs]: 16 to 512 µg/mL) and vancomycin (MICs: 64 to >1000 µg/mL). Vancomycin can induce expression of the VanA gene and has been identified in both Enterococcus faecium and Enterococcus faecalis. The VanB phenotype has also been identified in both E. faecium and E. faecalis and confers low-level resistance primarily to vancomycin. VanA, B, D, and E are all transferable to other organisms. In contrast, the VanC phenotypes are endogenous (constitutively produced) and are components of Enterococcus gallinarum, Enterococcus casseliflavus, and Enterococcus flavescens and confer resistance to vancomycin alone. The VanB gene has been identified in a strain of Streptococcus bovis. This gene showed 96% homology with the prototype VanB gene from E. faecalisV583, indicating the likelihood of the gene transfer from enterococcus to this strain of S. bovis.7
Vancomycin-intermediate S. aureus using the prior breakpoints of MIC 8 to 16 µg/mL was first reported in 1996 from Japan, and by June 2002, eight cases were confirmed in the United States.88 Using the new breakpoints, the incidence of vancomycin-intermediate S. aureus will increase. In June 2002, the first case of vancomycin-resistant S. aureus (MIC > 32 µg/mL) was identified in Michigan, followed in September 2002 by the second case in Pennsylvania.8,9 No mechanism of resistance has yet been identified from the strains of vancomycin-intermediate S. aureus, but the two strains of vancomycin-resistant S. aureus both possessed the VanA gene.
Tolerance is another mechanism by which bactericidal activity is decreased. Tolerance can be measured or assessed by two methods: the ratio of minimum bactericidal concentration to minimum inhibitory concentration (MBC : MIC) and time-kill curves. By definition, a MBC : MIC ratio of 32 or greater or less than 99.9% kill after 24 hours incubation in time-kill studies equates to tolerance. Tolerance to vancomycin has been identified in S. aureus, Streptococcus pneumoniae, and groups C and G streptococci.10–12
Spectrum of Activity
Vancomycin is active primarily against aerobic gram-positive cocci including Corynebacterium and methicillin-resistant S. aureus (MRSA). The MIC90 against methicillin-susceptible S. aureus (MSSA) is 1 µg/mL, and against MRSA it is 1 to 2 µg/mL.13–15 The incidence of vancomycin-intermediate or vancomycin-resistant S. aureus currently is very low and less than 1%. The activity of vancomycin against enterococci varies greatly with the species. E. faecium is the most resistant species of enterococci to vancomycin, with the resistant rates ranging from 30% to 90% depending on the institution. For all enterococci the vancomycin resistance rates are 20% to 25%.16
Most streptococci are susceptible to vancomycin, although it is considered an agent of last resort against these organisms. Vancomycin has been shown to be inferior to nafcillin or oxacillin for the treatment of MSSA infections. Treatment failures, prolonged treatment, and higher mortality rates have been demonstrated when vancomycin was used to treat MSSA infections compared with nafcillin or oxacillin.17,18
Vancomycin is active against anaerobic gram-positive organisms such as Peptostreptococcus spp., Propionibacterium spp., Eubacterium spp., Bifidobacterium spp., and most Clostridium spp., including C. difficile.19
Pharmacokinetics/Pharmacodynamics
Vancomycin is administered orally and intravenously (IV). The drug is poorly absorbed after oral administration, and the majority of the drug is excreted unchanged in feces. Inflammation of the gastrointestinal tract may result in increased absorption of vancomycin, and measurable serum concentrations might be obtained.20 Intramuscular injections are extremely painful and should not be used. Distribution of the drug is complete 1 hour after a 1- to 2-hour IV infusion. Vancomycin is approximately 55% bound to plasma proteins. The volume of distribution corrected for weight ranges from 0.4 to 0.9 L/kg.21–27 Vancomycin does not penetrate well into noninflamed meninges or aqueous humor.28 Distribution into inflamed meninges is variable, with reported ranges of 1% to 37% of serum concentrations29,30 and a mean concentration of 15% of serum or approximately 2.5 µg/mL.31 Penetration into ascitic, pericardial, and synovial fluids is greater than 75% serum concentrations; penetration approximates 50% into pleural fluid, and 30% to 50% into bile.25 Elimination of vancomycin is 80% to 90% unchanged drug in the urine via glomerular filtration and the remaining via nonrenal elimination. The nonrenal elimination rate in healthy individuals is 40 mL/min, and in chronic renal failure patients it is 6 mL/min.32 The half-life of the drug increases with decreased renal function; in patients with creatinine clearances (CrCl) greater than 80 mL/min, the half-life is 4 to 6 hours. The pharmacodynamic effect of vancomycin is time-dependent killing or time above the MIC.33 Therefore, the most important goal of therapy is to maintain a free serum trough concentration above the MIC of the organism. There is no documented correlation between serum peak concentrations and clinical outcomes.
Dosage Regimens
Oral Administration
Oral administration of vancomycin is only for treating C. difficile colitis and is considered second-line therapy for mild/moderate infections and primary therapy for moderate/severe infections. The dose is 125 to 500 mg orally every 6 hours and is not adjusted for renal dysfunction, owing to the poor absorption. Two oral formulations (capsules or liquid) can be used, or the IV solution can be administered orally to treat C. difficile. Table 124-1 lists dosing regimens for the antimicrobials discussed in this chapter.
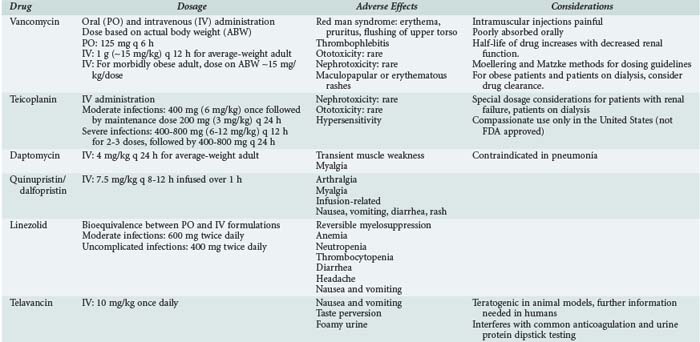
Intravenous Administration in Adults
In nonobese adults with normal renal function, the usual dose of vancomycin is 1 g (∼15 mg/kg) every 12 hours. This dose results in peak serum concentrations of 25 to 40 µg/mL 1 hour after completion of the infusion and trough serum concentration of 5 to 15 µg/mL. Dosing should be based on actual body weight. Several dosing guidelines have been developed to accurately and easily dose vancomycin. The most popular methods include the Moellering23 and Matzke24 nomograms. These methods use body weight and CrCl to calculate vancomycin dose. The weaknesses of these nomograms include the small number of patients used to develop and evaluate the nomogram and the fixed volume of distribution assumed for all patients (0.9 L/kg). Matzke24 found that for patients younger than 65 years of age, a volume of distribution of 0.7 L/kg may be more accurate, and for those older than 65, it is 0.9 L/kg. This variance in volume of distribution does affect the reproducibility of these nomograms when applied to different patient populations. The Cockcroft and Gault and modified Cockcroft and Gault methods of estimating CrCl are relatively reliable and accurate methods in patients of normal body mass.33
Morbidly obese patients are difficult to dose given the lack of pharmacokinetic studies. Doses of approximately 30 mg/kg/d based on actual body weight should provide a peak serum concentration of 25 to 35 µg/mL. Because CrCl is the best correlate to vancomycin clearance, the most accurate method for estimating CrCl should be used and varies with body mass. CrCl estimations in the obese patient are best predicted by the Salazar-Corcoran method.34 Young obese patients with no comorbid conditions affecting renal function often require the dosing interval to be more frequent to achieve a trough serum concentration of 5 to 15 µg/mL. This is due to the faster rate of clearance of the drug (2.3-2.5 times higher) in obese compared with nonobese patients.35,36
Vancomycin Dosing in Critically Ill Patients
Garaud evaluated critically ill patients and found an average volume of distribution of 0.6 L/kg in patients with CrCl greater than 70 mL/min and 0.4 L/kg with CrCl of 10 to 60 mL/min.26 Critically ill patients are often receiving medications to improve hemodynamics, such as dopamine, dobutamine, and furosemide. These medications result in increases in renal function and changes in volume status for the patient. In a study designed to assess the impact of such medications on vancomycin pharmacokinetics, two observations were made.37 First, some of the patients required larger total daily doses of vancomycin to achieve therapeutic concentrations than the Moellering nomogram predicted (26.78 + 3.01 mg/kg/d versus 18.95 + 3.41 mg/kg/d). Second, on discontinuation of these medications, the serum trough concentrations increased despite no change in CrCl or body weight. The theory is that these medications enhanced vancomycin clearance by improving renal blood flow and/or interacting with the renal anion transport system, thus increasing glomerular filtration and renal tubular secretion. Therefore, larger doses of vancomycin may be required while on these medications, and smaller doses may be more appropriate on discontinuation of these medications.
Vancomycin Dosing for Patients on Dialysis/Hemofiltration/Cardiopulmonary Bypass
The percentage of vancomycin removed by low-permeability cellulose hemodialyzers is 4% to 6.9%.38–40 Therefore, no supplemental vancomycin dosing is required after hemodialysis with these older systems. The removal of vancomycin during intradialytic administration has been studied using three types of cellulose membranes: cellulose acetate (CA), cellulose triacetate (CT), and CA high-performance 210 (CAHP-210). With the CA membranes, 0% to 25% (mean of ∼13%) of vancomycin is removed.38,41 The CT membranes remove 16% to 44% (mean of ∼26%) of vancomycin.38,41 Vancomycin removal during intradialytic administration with the CAHP-210 membranes is 0% to 35%, with a mean of 24%.42 High-flux synthetic membranes such as polysulfone or polyacrylonitrile remove significantly more vancomycin than do the cellulose membranes, with 30% to 55% and 25% to 40% of vancomycin removed, respectively.38–40,43–45
Continuous renal replacement therapy (CRRT) is a low-volume (1-2 L/h) therapy. The most frequently used methods of CRRT are continuous venovenous hemofiltration (CVVH), continuous venovenous hemodialysis (CVVHD), and continuous arteriovenous hemodialysis (CAVHD). Both CVVHD and CAVHD result in a greater total body clearance of vancomycin than does hemofiltration. The clearances achieved with each of these methods vary with blood flow rate, ultrafiltration rate, and the membranes used. The total clearance of vancomycin with CVVHD or CAVHD is 31 to 39 mL/min, and the half-life ranges from 14 to 25 hours.46–49 Clearance of vancomycin in patients with normal renal function (CrCl > 70 mL/min) and with mild renal dysfunction (CrCl 40-70 mL/min) has been reported to be 88 and 48 mL/min, respectively.50 High-volume hemofiltration (HVHF), with an ultrafiltration rate of 6 L/h, increases vancomycin clearance to approximately 60 mL/min.51 Therefore, patients receiving CAVHD or CVVHD should receive vancomycin every 36 to 48 hours, and those undergoing HVHF should receive the drug every 12 to 24 hours.
Cardiopulmonary bypass (CPB) significantly impacts the pharmacokinetic parameters of vancomycin. Immediately after initiating CPB, vancomycin serum concentration decreased by 7 µg/mL (5.7 to 8.4 µg/mL), which represented approximately a 38% decrease in concentration.52 Over the next 30 minutes, serum vancomycin concentration may increase 1 to 2 µg/mL but thereafter gradually and steadily decreases.52 The half-life is not affected by CPB and does not change during the process.
Adverse Effects
Common toxicities that have been associated with vancomycin therapy include red man syndrome, thrombophlebitis, ototoxicity, and nephrotoxicity. Evidence establishing a clear relationship between these toxicities and vancomycin peak or trough concentrations or the incidence of these events is limited and contradictory.4,53–55
Red man syndrome comprises erythema, pruritus, and flushing of the upper torso and is often associated with too rapid an infusion of the drug. In general, the infusion rate should not exceed 1 g/h. Less frequently, hypotension and angioedema can occur. It is thought that increased histamine release is the cause of this syndrome.4,54–56 A comparative trial of once-daily versus twice-daily vancomycin found the incidence of this syndrome to be 13.7% and 9.6%, respectively.54 The effects of red man syndrome can be relieved by antihistamines.57,58
Thrombophlebitis is reported in 3% to 23% of patients receiving vancomycin and is more common in patients who receive vancomycin for more than 7 days or have peripheral catheter lines for prolonged durations.4,54
Ototoxicity rates range from 0% to 9% in patients receiving vancomycin.4,54 The definition of ototoxicity ranges from tinnitus to hearing loss. The evidence demonstrating any relationship between ototoxicity and high peak serum concentrations of vancomycin is limited. In cancer patients, only 4 of 19 patients with ototoxicity had elevated serum concentrations of vancomycin, and only 1 had a concentration greater than 80 µg/mL.4 Others have reported ototoxicity associated with peak serum concentrations of 37.5 to 152 µg/mL.59,60 A trial comparing once-daily to twice-daily dosing of vancomycin demonstrated more frequent ototoxicity in the twice-daily dosed group (15.6% versus 3.2%), which had a significantly lower peak concentration and similar trough concentration compared to the group receiving daily doses.54 This lack of correlation between serum concentrations of vancomycin and ototoxicity suggests that the observed toxicity was due to either another drug or to the combination of another drug with vancomycin. In the majority of cases, ototoxicity symptoms disappear within a month of discontinuing vancomycin.
The issue of nephrotoxicity associated with vancomycin is complicated by several confounding factors. The original formulation was very impure, and the impurities were associated with toxicities including nephrotoxicity. In addition, many definitions of nephrotoxicity have been used over the years, different patient populations have been studied, and different doses used, making it difficult to compare one study to another. In general, the rate of nephrotoxicity is 5% to 10% when vancomycin is not administered with other nephrotoxic agents and trough concentrations are less than 10 µg/mL.54,61,62 Elting and colleagues identified older age, Acute Physiology and Chronic Health Evaluation (APACHE) score greater than 40, and duration of therapy of greater than 14 days to be the best predictors for a patient to develop nephrotoxicity due solely to vancomycin therapy.4 A number of other studies have found an increased incidence of nephrotoxicity (21%-35%) when vancomycin serum trough concentrations are greater than 10 µg/mL.62–64 In addition, Lodise demonstrated an increased rate of nephrotoxicity (∼35%) when the total daily dose is 4 grams or more compared to total doses less than 4 grams (∼11%).65 Studies have demonstrated higher rates of nephrotoxicity when vancomycin is used in combination with an aminoglycoside compared with either agent alone.62,66,67 Goetz performed a meta-analysis of eight studies and found the incidence of nephrotoxicity associated with combination therapy was 13% greater than with vancomycin alone and 4% greater than with an aminoglycoside alone.67
Other toxicities associated with vancomycin include maculopapular or erythematous rashes (2%-8%)26,68,75 and anecdotal reports of neutropenia and thrombocytopenia.68,69
Therapeutic Drug Monitoring
Routine monitoring of vancomycin serum concentrations has become a highly debated issue over the years. Those who advocate routine monitoring cite the need to ensure therapeutic concentrations as well as minimize toxicities. To date there is only one trial that compared efficacy and toxicity with high-dose once-daily versus twice-daily dosing of vancomycin; with these dosing regimens, peak serum concentrations were vastly different but trough serum concentrations were similar.54 The mean peak serum concentrations in the once-daily and twice-daily dosed groups were 42.8 + 16.1 and 27.0 + 9.2 µg/mL, respectively. There were no differences in clinical efficacy, red man syndrome, thrombophlebitis, ototoxicity, and nephrotoxicity between the two groups.
Studies over the past 20 years have shown that peak concentrations of vancomycin are not associated with toxicities or clinical efficacy. Therefore, monitoring peak serum concentrations only adds to hospital and healthcare system costs and provides no beneficial clinical information. Some studies have demonstrated a correlation of nephrotoxicity to serum trough concentrations ≥ 10 µg/mL, whereas others have not. Given the lack of consensus, it may be prudent to measure serum trough concentrations until more definitive studies are conducted to address this issue.70
In patients with end-stage renal disease, the fluorescence polarization immunoassay (FPIA) overestimates vancomycin concentrations.71 FPIA is the most common method for determining vancomycin concentrations, and when it was compared with the enzyme multiplied immunoassay technique, it was found to produce higher peak serum concentrations by 7 to 11 µg/mL and higher trough concentrations by 4 to 6 µg/mL.
 Teicoplanin
Teicoplanin
Mechanisms of Action And Resistance
Teicoplanin, like other glycopeptide antibiotics, inhibits synthesis of the cell wall by binding to the D-alanyl-D-alanine terminus of cell wall precursor units. Resistance has been reported in both staphylococci and enterococci. The VanA phenotype confers high-level resistance to both teicoplanin (MIC: 16 to 512 µg/mL) and vancomycin (MIC: 64 to >1000 µg/mL). The VanB phenotype has also been identified in both E. faecium and E. faecalis and usually confers low-level resistance to vancomycin but not to teicoplanin. This may limit the utility of teicoplanin for some vancomycin-resistant enterococcal infections. Several reports of S. aureus resistance developing during therapy with teicoplanin have been reported.72–74 The mechanism of the resistance was determined in one patient to be constitutive and non-plasmid mediated.73
Spectrum of Activity
Teicoplanin is only active against gram-positive organisms. Activity against MSSA and MRSA is comparable to that of vancomycin. Coagulase-negative staphylococci have a varied pattern of susceptibility to teicoplanin. Staphylococcus haemolyticus is the most resistant species to teicoplanin (30%).75 These isolates are 25% more resistant to teicoplanin than to vancomycin. Against methicillin-resistant coagulase-negative staphylococci, 39% of isolates have teicoplanin MICs greater than 8 µg/mL compared with 1% with vancomycin.75,76 Teicoplanin is similar in activity to vancomycin against enterococci, although its reliability in treating infections with VanB resistance to vancomycin may be limited. Teicoplanin is active against other aerobic and anaerobic gram-positive organisms such as Corynebacterium spp., Clostridium spp., including C. difficile and C. perfringens, Peptostreptococcus spp., and Propionibacterium acnes.
Pharmacokinetics/Pharmacodynamics
Teicoplanin is administered orally and intravenously. The drug is poorly absorbed after oral administration, and approximately 40% of the drug is excreted unchanged in feces. The pharmacokinetic model that best describes the elimination of teicoplanin is triexponential. IV administration of 400 mg (6 mg/kg) should provide a peak serum concentration of 20 to 50 µg/mL attained 1 hour after administration.77 The volume of distribution is large at 0.9 to 1.41 L/kg, and teicoplanin is 90% to 95% protein bound.77 Penetration into body fluids and tissues has not been extensively studied. Penetration into noninflamed meninges and fat is poor, but distribution into myocardium and pericardium is good.78,79 Teicoplanin is primarily eliminated via glomerular filtration, and only 3% is metabolized.77 The half-life is approximately 150 hours in patients with normal renal function.77 Because of the long half-life, it takes 14 days to reach steady state. In patients with CrCl of 13 to 25, the half-life was found to be 280 to 667 hours.80,81
Dosage Regimens/Therapeutic Drug Monitoring
Despite the long half-life in patients with normal renal function, teicoplanin should be administered daily, and the dose is dependent on the severity of infection. For less serious infections involving the urinary tract, skin, soft tissue, and lower respiratory tract, a loading dose of 400 mg (6 mg/kg) × 1 is administered, followed by a maintenance dose of 200 mg (3 mg/kg) every 24 hours. For severe infections such as septicemia, endocarditis, and osteomyelitis, 400 mg of teicoplanin is administered every 12 hours for 3 doses, followed by 400 mg every 24 hours.77 Although no therapeutic range has been established for teicoplanin, trough concentrations should be at least 10 µg/mL.77
Renal Failure/Dialysis
Teicoplanin is not removed by hemodialysis or continuous ambulatory peritoneal dialysis (CAPD).82,83 The amount removed by CVVHD is dependent on the flow rate but is often minimal.84,85 Several dosing regimens exist for renal dysfunction, and the simplest method is administering a dose of 6-10 mg/kg every 48 to 72 hours.
Adverse Effects
Nephrotoxicity associated with teicoplanin is much lower than with vancomycin. The incidence from published and unpublished studies found the nephrotoxic rate to be 4%.55 Ototoxic rates with teicoplanin are similar to those with vancomycin.55 Hypersensitivity reactions are the most common adverse reaction to teicoplanin (2%-15%).55
 Daptomycin
Daptomycin
Mechanisms of Action And Resistance
Daptomycin has a unique mechanism of action and has been found to inhibit lipoteichoic acid synthesis, owing to binding to the membrane in the presence of calcium.86,87 Minimal information is available on the mechanism(s) of resistance to daptomycin. Limited in vitro studies have been performed attempting to create daptomycin resistance in the laboratory.88 Mechanisms of resistance have not been elucidated, and the clinical relevance of in vitro resistance is unknown.
Spectrum of Activity
Daptomycin’s antibacterial activity encompasses most gram-positive bacteria, including vancomycin-resistant isolates and penicillin-resistant pneumococci. The MICs of daptomycin are 8- to 16-fold lower in the presence of calcium. Therefore, all in vitro testing must be supplemented with physiologic concentrations of calcium.89 The breakpoint for susceptible is ≤1 µg/mL for staphylococci and β-hemolytic streptococci. Given the rare number of isolates not susceptible to daptomycin, a resistant breakpoint has yet to be determined. The MIC90 against MSSA, MRSA, Staphylococcus epidermidis, and Staphylococcus saprophyticus are all 0.5 µg/mL or less.89,90 In a recent surveillance study, 7 S. aureus and 6 coagulase-negative staphylococci were non-susceptible to daptomycin.91 Daptomycin also appears active against vancomycin-intermediate and vancomycin-resistant strains of S. aureus.92,93 The breakpoint for susceptible against enterococci is ≤4 µg/mL, and again no resistant breakpoint has been established. Against E. faecalis and E. faecium, including vancomycin-resistant strains, the MIC90 is 2 µg/mL or less.89,90 Daptomycin resistance is higher among E. faecium than E. faecalis.91 The MIC90 is 0.25 µg/mL against S. pneumoniae and β-hemolytic streptococci, and resistance has not been reported with these organisms.89–91
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


