12 Acid-Base Disorders
 General Principles
General Principles
Three widely accepted methods are used to analyze and classify acid-base disorders, yielding mutually compatible results. The approaches differ only in assessment of the metabolic component (i.e., all three treat PCO2 as an independent variable): (1) HCO3− concentration ([HCO3−]); (2) standard base-excess; (3) strong ion difference. All three yield virtually identical results when used to quantify the acid-base status of a given blood sample.1–4 For the most part, the differences among these three approaches are conceptual; in other words, they differ in how they approach the understanding of mechanism.5–7
There are three mathematically independent determinants of blood pH:
 Assessing Acid-Base Balance
Assessing Acid-Base Balance
 Metabolic Acid-Base Disorders
Metabolic Acid-Base Disorders
Pathophysiology of Metabolic Acid-Base Disorders
Disorders of metabolic acid-base balance occur as a result of:
The Kidneys
Normal plasma flow to the kidneys is approximately 600 mL/min in adults. The glomeruli filter the plasma to yield about 120 mL/min of filtrate. Normally, more than 99% of the filtrate is reabsorbed and returned to the plasma. Thus, the kidney can only excrete a very small amount of strong ions into the urine each minute, and several minutes to hours are required to achieve a significant impact on SID. The handling of strong ions by the kidney is extremely important because every Cl− ion that is filtered but not reabsorbed decreases SID. Accordingly, “acid handling” by the kidney is generally mediated through changes in Cl− balance. The purpose of renal ammoniagenesis is to allow the excretion of Cl− without Na+ or K+. Viewed this way, renal tubular acidosis can be regarded as an abnormality of Cl− handling rather than of H+ or HCO3− handling.3
Renal-Hepatic Interaction
Ammonium ion (NH4+) is important to systemic acid-base balance not because it stores H+ or has a direct action in the plasma (normal plasma NH4+ concentration is <0.01 mEq/L). NH4+ is important because it is “co-excreted” with Cl−. Of course, NH4+ is not only produced in the kidney. Hepatic ammoniagenesis (and, as we shall see, glutaminogenesis) is also important for systemic acid-base balance and is tightly controlled by mechanisms sensitive to plasma pH.8 This reinterpretation of the role of NH4+ in acid-base balance is supported by the evidence that hepatic glutaminogenesis is stimulated by acidosis.9 Glutamine is used by the kidney to generate NH4+ and thus facilitates the excretion of Cl−. The production of glutamine, therefore, can be seen as having an alkalinizing effect on plasma pH because of the way the kidney utilizes it.
The Gastrointestinal Tract
Different parts of the GI tract handle strong ions in distinct ways. In the stomach, Cl− is pumped out of the plasma and into the lumen, thereby reducing the SID and pH of gastric juice. The pumping action of the gastric parietal cells increases SID of the plasma by promoting the loss of Cl−; this effect produces the so-called alkaline tide at the beginning of a meal when gastric acid secretion is maximal.10 In the duodenum, Cl− is reabsorbed and the plasma pH is restored. Normally, only slight changes in plasma pH are evident because Cl− is returned to the circulation almost as soon as it is removed. However, if gastric secretions are removed from the patient, either through a suction catheter or as a result of vomiting, Cl− is lost and SID increases. It is important to realize that it is the Cl− loss, not the H+ loss, that is the cause for widening of the SID and the development of metabolic alkalosis. Although H+ is “lost” as HCl, it is also lost with every molecule of water removed from the body.
 Metabolic Acidosis
Metabolic Acidosis
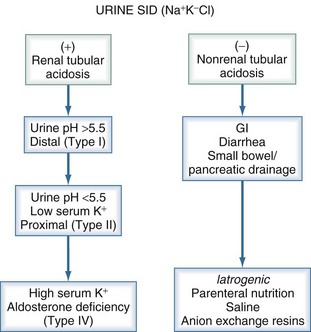
Traditionally, metabolic acidoses are categorized according to the presence or absence of unmeasured anions. The presence of unmeasured anions is routinely inferred by measuring the concentrations of electrolytes in plasma and calculating the anion gap, as described later. The differential diagnosis for a positive–anion gap (AG) acidosis is shown in Box 12-1. Non–anion gap acidoses can be divided into three types: renal, GI, and iatrogenic (Figure 12-1). In the intensive care unit (ICU), the most common types of metabolic acidosis include lactic acidosis, ketoacidosis, iatrogenic acidosis, and acidosis secondary to toxins.
The potential effects of metabolic acidosis and alkalosis on vital organ function are shown in Table 12-1. Metabolic and respiratory acidosis may have different implications with respect to survival, an observation that suggests that the underlying disorder is perhaps more important than the absolute degree of acidemia.11
TABLE 12-1 Potential Clinical Effects of Metabolic Acid-Base Disorders
| Metabolic Acidosis | Metabolic Alkalosis |
|---|---|
| Cardiovascular | Cardiovascular |
| Decreased inotropy | Decreased inotropy (Ca++ entry) |
| Conduction defects | Altered coronary blood flow* |
| Arterial vasodilation | Digoxin toxicity |
| Venous vasoconstriction | |
| Oxygen Delivery | Neuromuscular |
| Decreased oxy-Hb binding | Neuromuscular excitability |
| Decreased 2,3-DPG (late) | Encephalopathy seizures |
| Neuromuscular | Metabolic Effects |
| Respiratory depression | Hypokalemia |
| Decreased sensorium | Hypocalcemia |
| Hypophosphatemia | |
| Impaired enzyme function | |
| Metabolism | Oxygen Delivery |
| Protein wasting | Increased oxy-Hb affinity |
| Bone demineralization | Increased 2,3-DPG (delayed) |
| Catecholamine, PTH, and aldosterone stimulation | |
| Insulin resistance | |
| Free radical formation | |
| Gastrointestinal | |
| Emesis | |
| Gut barrier dysfunction | |
| Electrolytes | |
| Hyperkalemia | |
| Hypercalcemia | |
| Hyperuricemia |
2,3-DPG, 2,3-diphosphoglycerate; oxy-Hb, oxyhemoglobin; PTH, parathyroid hormone.
* Animal studies have shown both increased and decreased coronary artery blood flow.
If metabolic acidemia is to be treated, consideration should be given to the likely duration of the disorder. If it is expected to be short lived (e.g., diabetic ketoacidosis), maximizing respiratory compensation is usually the safest approach. Once the disorder resolves, ventilation can be quickly reduced to normal, and there will be no lingering effects of therapy. However, if the disorder is likely to be more chronic (e.g., renal failure), therapy aimed at restoring SID is indicated. In all cases, the therapeutic target can be quite accurately determined from the standard base-excess. As discussed, the standard base-excess corresponds to the amount SID must change in order to restore the pH to 7.4, assuming a PCO2 of 40 mm Hg. Thus, if the SID is 30 mEq/L and the standard base-excess is −10 mEq/L, the target SID would be 40 mEq/L. Accordingly, the plasma Na+ concentration would have to increase by 10 mEq/L for NaHCO3 administration to completely repair the acidosis. If increasing the plasma Na+ concentration is inadvisable for other reasons (e.g., hypernatremia), then NaHCO3 administration is also inadvisable. Importantly, NaHCO3 administration has not been shown to improve outcome in patients with lactic acidosis.12
In addition, NaHCO3 administration is associated with certain disadvantages. Large (hypertonic) doses given rapidly can lead to hypotension13 and have the potential to cause a sudden marked increase in PaCO2.14 Accordingly, it is important to assess the patient’s ventilatory status before NaHCO3 is administered, particularly in the absence of mechanical ventilation. NaHCO3 infusion also affects circulating [K+] and [Ca++] concentrations, which need to be monitored closely.
Tromethamine (Tris-buffer or Tham) is an organic buffer that readily penetrates cells.15 It is a weak base (pK = 7.9) that does not alter SID and does not affect plasma [Na+]. Accordingly, it is often used when administration of NaHCO3 is contraindicated because of hypernatremia. This agent has been available since the 1960s, but limited data are available on its use in humans with acid-base disorders. In small uncontrolled studies, tromethamine appears to be effective in reversing metabolic acidosis secondary to ketoacidosis or renal failure without obvious toxicity.16 However, adverse reactions have been reported, including hypoglycemia, respiratory depression, and even fatal hepatic necrosis when concentrations exceeding 0.3 M are used. In Europe, a mixture of tromethamine, acetate, NaHCO3, and disodium phosphate is available (Tribonate). This mixture seems to have fewer side effects than tromethamine alone, but experience with Tribonate is still quite limited.
Anion Gap and Strong Ion Gap
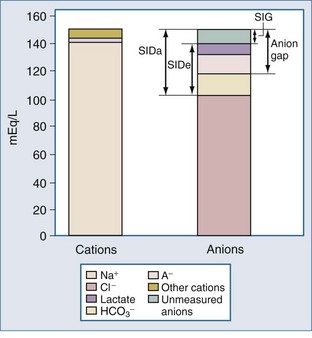
For more than 30 years, AG has been used by clinicians, and it has evolved into a major tool to evaluate acid-base disorders.17 AG is estimated from the differences between the routinely measured concentrations of serum cations (Na+ and K+) and anions (Cl− and HCO3−). Normally this difference, or “gap,” is made up by albumin and, to a lesser extent, by phosphate. Sulfate and lactate also contribute a small amount, normally less than 2 mEq/L. However, there are also unmeasured cations, such as Ca++ and Mg++, and these tend to offset the effects of sulfate and lactate, except when the concentration of sulfate or lactate is abnormally increased (Figure 12-2). Plasma proteins other than albumin can be positively or negatively charged, but in the aggregate tend to be neutral except in rare cases of abnormal paraproteins, such as in cases of multiple myeloma.18 In practice, AG is calculated as follows:
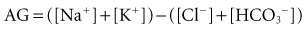
Because of its low and narrow extracellular concentration range, K+ is often omitted from the calculation. The normal value for AG is 12 ± 4 (if [K+] is considered) or 8 ± 4 mEq/L (if [K+] is not considered). The normal range has decreased in recent years following the introduction of more accurate methods for measuring Cl− concentration.19,20 However, the various measurement techniques available mandate that each institution reports its own expected “normal anion gap.”
The AG is useful because this parameter can limit the differential diagnosis for patients with metabolic acidosis. If AG is increased, the explanation almost invariably will be found among five disorders: ketosis, lactic acidosis, poisoning, renal failure, or sepsis.21 However, several conditions can alter the accuracy of AG estimation, and these conditions are particularly prevalent among patients with critical illness22,23:
Other factors that can increase AG are low Mg++ concentration and administration of the sodium salts of poorly reabsorbable anions (e.g., beta-lactam antibiotics).25 Certain parenteral nutrition formulations, such as those containing acetate, can increase AG. Citrate-based anticoagulants rarely can have the same effect after administration of multiple blood transfusions.26 None of these rare causes, however, increases AG significantly,27 and they are usually easily identified. In recent years, some additional causes of an increased AG have been reported. It is sometimes widened in patients with nonketotic hyperosmolar states induced by diabetes mellitus; the biochemical basis for this effect remains unexplained.28 In recent years, unmeasured anions have been reported in the blood of patients with sepsis29,30 and liver disease31,32 and in experimental animals injected with endotoxin.33 These anions may be the source of much of the unexplained acidosis seen in patients with critical illness.34
Additional doubt has been cast on the diagnostic value of AG in certain situations, however.22,30 Salem and Mujais22 found routine reliance on AG to be “fraught with numerous pitfalls.” The primary problem with the AG is its reliance on the use of a “normal” range that depends on normal circulating levels of albumin and to a lesser extent phosphate, as discussed earlier. Plasma concentrations of albumin or phosphate are often grossly abnormal in patients with critical illness, leading to changes in the “normal” range for AG. Moreover, because these anions are not strong anions, their charge is affected by pH.
These considerations have prompted some authors to adjust the “normal range” for AG according to the albumin concentration24 or phosphate concentration.6 Each g/dL of albumin has a charge of 2.8 mEq/L at pH 7.4 (2.3 mEq/L at pH 7.0 and 3.0 mEq/L at pH 7.6). Each mg/dL of phosphate has a charge of 0.59 mEq/L at pH 7.4 (0.55 mEq/L at pH 7.0 and 0.61 mEq/L at pH 7.6). Thus, the “normal” AG can be estimated using this formula6:
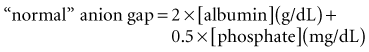
Or for international units:
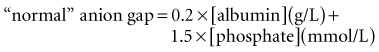
These formulas only should be used when the pH is less than 7.35, and even then they are only accurate within 5 mEq/L. When more accuracy is needed, a slightly more complicated method of estimating [A−] is required.31,35
Another alternative to using the traditional AG is to use the SID. By definition, SID must be equal and opposite to the negative charges contributed by [A−] and total CO2. The sum of the charges from [A−] and total CO2 concentration has been termed the effective strong ion difference (SIDe).18 The apparent strong ion difference (SIDa) is obtained by measurement of each individual ion. Both the SIDa and the SIDe should equal the true strong ion difference. If the SIDa and SIDe differ, unmeasured ions must exist. If the SIDa is greater than SIDe, these ions are anions; if the SIDa is less than SIDe, the unmeasured ions are cations. This difference has been termed the strong ion gap to distinguish it from AG.31 Unlike the AG, the strong ion gap is normally zero and does not change with changes in pH or albumin concentration.
Positive–Anion Gap Acidoses
Lactic Acidosis
In many forms of critical illness, lactate is the most important cause of metabolic acidosis.36 Blood lactate concentration has been shown to correlate with outcome in patients with hemorrhagic37 and septic shock.38 Lactic acid has been viewed as the predominant source of metabolic acidosis due to sepsis.39 In this view, lactic acid is released primarily from the musculature and the gut as a consequence of tissue hypoxia. Moreover, the amount of lactate produced is believed to correlate with the total oxygen debt, the magnitude of hypoperfusion, and the severity of shock.36 In recent years, this view has been challenged by the observation that during sepsis, even with profound shock, resting muscle does not produce lactate. Indeed, studies by various investigators have shown that the musculature actually may consume lactate during endotoxemia.40–42 Data concerning the gut are less clear. There is little question that underperfused gut can release lactate; however, it does not appear that the gut releases lactate during sepsis if mesenteric perfusion is maintained. Under such conditions, the mesenteric circulation can even become a net consumer of lactate.40,41 Perfusion is likely to be a major determinant of mesenteric lactate metabolism. In a canine model of sepsis, gut lactate production could not be shown when flow was maintained with dopexamine hydrochloride.42
Studies in animals as well as humans have shown that the lung may be a prominent source of lactate in the setting of acute lung injury.40,43–45 While studies such as these do not address the underlying pathophysiologic mechanisms of hyperlactatemia in sepsis, they suggest that using blood lactate concentration as evidence for tissue dysoxia is an oversimplification at best. Indeed, many investigators have begun to offer alternative interpretations of hyperlactatemia in this setting.44–48 Box 12-2 lists several alternative sources of hyperlactatemia. In particular, pyruvate dehydrogenase, the enzyme responsible for moving pyruvate into the Krebs cycle, is inhibited by endotoxemia.49 However, data from recent studies suggest that increased aerobic metabolism may be more important than metabolic defects or anaerobic metabolism.50 Finally, administration of epinephrine promotes lactic acidosis, presumably by stimulating cellular metabolism (e.g., increased glycolysis in skeletal muscle).





