Relaxants
Reversal Drugs
Alcuronium
Diadonium
PANCURONIUM
NEOSTIGMINE
Amidonium
Dipyrandium
Pipecuronium
Edrophonium
Anatruxonium
Duador
Quilidium
Pyridostigmine
ATRACURIUM
Fazadinium
Rapacuronium
Galanthamine
Benzoquinonium
Laudexium
ROCURONIUM
4-aminopyridine
C-Toxiferin
Maluetin
Stercuronium
3:4-diaminopyridine
CISATRACURIUM
Metocurine
SUCCINYLCHOLINE
SUGAMMADEX
Cycobutonium
MIVACURIUM
Tercuronium
Dacuronium
Myanesin
Toxiferine
Decadonium
Mytolon
Tubocurarine
Decamethonium
N-methyl Tubocurarine
VECURONIUM
Doxacurium
Nubitanium
In the Beginning
“The natives poisoned their arrows with the juice of a death-dealing herb. In a twinkling the forty-seven Spaniards were pierced with arrow-wounds, before they could protect themselves with their shields. There was but one man who survived, all the rest perishing from the effects of the poison.”
Thus was the existence of a paralyzing drug revealed to Europeans by Pieter Martyr D’Anghere in his fifteenth century treatise De Orbe Novo. Rather than traveling with the conquistadores like embedded modern day correspondents, D’Anghere simply interviewed those returning from the “New World.” A century later, in 1594, Sir Walter Raleigh returned from an incursion into now Venezuela and described a tribe he encountered, the Aroras, as “people who used strong poison in their arrows.” Lawrence Keynes, one of Raleigh’s lieutenants, named this strong poison “ourari”, literally the “bird-killer.” One can imagine the transliteration of ourari into curare.
Two plant groups, Chondodendron Tomentosum and Strychnos Toxifera supplied the poison. The ritual preparation of the poison took several days. An apocryphal story has it that some tribes sequestered old women with the necessary knowledge and skills throughout the process of manufacture. The resulting condition of the women defined the potency of the tarry substance produced. If they still stood and appeared relatively healthy, the batch was deemed insufficiently potent and discarded. In contrast, if the women were weak, unable to stand, the poison was deemed acceptable. Given these working conditions, it is surprising that any of these women lived to become old and experienced. Having passed this first potency test, a frog was pricked with the poison, and the number of leaps the frog then made, before expiring, quantified the potency. The laborious process, and the limited availability of old women, made the toxin valuable. Its use was commonly restricted to hunting and not warfare.
Although von Humboldt in one place in his book partially confirms the above apocryphal story in which old women made and were the test subjects for establishment of potency of the curare preparation, in another place he denies that such testing could be possible [2]. Charles Waterton adds to the latter view, stating that women and young girls were excluded from the preparation of the curare concentrate [2]. Given the highly ionized nature of curare and other muscle relaxants, it is difficult to imagine how curare could be vaporized. If this is correct, then inhalation of curare in the vapors made in the process of purification seems unlikely.
Over the next four centuries, more reports and crucial observations were made. The poison was not toxic if ingested, inhaled, or applied directly to or injected into a nerve [3]. Also, after poisoning with curare, the heart continued to beat, and artificial respiration saved the animal from death [4,5].
Eighteenth and Nineteenth Centuries: Early Scientific Investigations
Explorers Discover and Bring Back Curare
Explorer-scientists, including Charles-Marie De La Condamine, Edwin Bancroft, and Charles Waterton, travelled extensively in different areas of South America. They provided the raw material to European pharmacologists and physiologists who undertook the initial studies of curare. These hardy explorers often put themselves at personal risk; the Amazonian forest was an inhospitable place, and working with the raw material was dangerous. The French scientist FD Herissant describes how he and his assistant became weak and almost unconscious while boiling the crude curare extract in order to create a concentrated mixture. “A pint of good wine and sugar” cured both investigators.
The eighteenth century physician, Edwin Bancroft, brought back samples of crude curare from South America for study. He was the first to thoroughly report the diverse tribal methods of preparation and storage of the curare. The natives used a variety of vessels for storage of the curare preparation: a clay pot (pot curare), a calabash gourd (calabash curare), or a hollow piece of bamboo (tube curare, hence “tubocurare”). His son, Edward Bancroft provided samples to the English surgeon, Benjamin Brodie, who made the crucial observation that animals poisoned with curare could be kept alive by inflating their lungs [5]. Brodie also suggested treatment of tetanus with curare [6].
1814
No early explorer was more colorful than Charles Waterton, an English nobleman who managed his family’s estates in South America. His exploits included scaling the dome of St. Peter’s Cathedral in Rome, much to the displeasure of the Pope. While in Guiana, he showed that curare was effective in many species, including large animals. He witnessed Brodie and William Sewell’s famous 1814 public demonstration of the effect of curare on a donkey.
“A she ass received the wourali poison in the shoulder and died in apparently 10 minutes. An incision was made in its windpipe, and through it the lungs were regularly inflated for two hours with a pair of bellows. Suspended animation returned. The ass held up her head and looked around.”
The ass “now named Wouralia” was put out to graze at Walton Hall, on the estate of Earl Percy, who stated, “No burden shall be placed on her and she shall end her days in peace.” Wouralia lived on in this happy state for almost 25 more years [7].
1838
Twenty years after the “Wouralia” demonstration, Sewell, a veterinary surgeon, used curare to treat two horses with tetanus. Both animals died. Despite this failure, curare was later used in humans to treat disorders involving spasticity. None proved successful.
1846
In 1846, the French polymath Claude Bernard (1813–1878) discovered the site of action of curare (Fig. 50.1) [8]. In curarized (paralyzed) frogs, he noted that electrical impulses still passed down the motor nerves [8], and that electrical impulses applied directly to the muscle caused contraction. He concluded that curare acted where the nerve impulse reached the muscle, the neuromuscular junction. It was not until the 1930s that Henry Dale and others showed that acetylcholine released from the motor nerve terminal effected neuromuscular transmission, and curare acted by blocking acetylcholine receptors.

Fig. 50.1
Claude Bernard in his laboratory. The drawing shows the equipment he used to demonstrate the site of action of curare at the neuromuscular junction. (Courtesy of Wellcome Images UK.)
1858
The surgeon Lewis Sayre, and his house officer FA Burrell, at Bellevue Hospital New York, took up Brodie’s suggestion that curare might be used to treat tetanus. In a patient with tetanus, the standard treatments of brandy, beef tea, (by both mouth and per rectum) had failed. Four applications of a dilute solution of the “woorari” poison were applied directly onto the hand wound through which the original infection occurred. The patient died during a severe spasm shortly thereafter [9]. Despite such setbacks, the search for a therapeutic use of curare in spastic conditions such as chorea, rabies, epilepsy and strychnine poisoning continued. But curare extract sufficient to decrease spasticity inevitably produced fatal respiratory failure.
1860s
The Edinburgh scientists Thomas Fraser and Alexander Brown made the next breakthrough demonstrating that conversion of alkaloids with tertiary (three atom) nitrogen groups (e.g., atropine or morphine) to a quaternary (four atom) form produced compounds with curare-like activity [10]. This first demonstration of a structure-activity relationship in medicine was fundamental to drug development a century later.
1900–1940: Some False Starts
First Medical Trials with Curare
Despite poor therapeutic experiences, many considered using the curare “poison” for medical purposes, but its inherent dangers made it unsuitable as a drug fortrial and error experimentation. Nonetheless, we were getting nearer to the discovery of the beneficial uses of curare.
1906
In 1906, the chemist August von Hofmann synthesized quaternary amines (Fig. 50.2). Muscle relaxants have this structure [11]. He also described the capacity of increased pH and temperature to break down a quaternary amine to a tertiary amine, the eponymous Hofmann reaction. More than seventy years later, John Stenlake (Fig. 50.2) at the University of Strathclyde in Glasgow made use of this reaction in the design of the then revolutionary drug, atracurium [12].

Fig. 50.2
Two scientists at either end of the Hofmann elimination story. August Hofmann (left) described the reaction that bears his name, in 1907 (photograph from fineartamerica.com). John Stenlake (right) used the reaction described by Hofmann in the development of atracurium more than 70 years later (1981). (Image of August Hoffman is courtesy of Wellcome Images UK.)
Also in 1906, the pharmacologists R Hunt and R Taveau synthesized succinylcholine and studied it in animals [13]. However, their anesthetized animals had already been paralyzed with curare, and thus they did not observe succinylcholine’s neuromuscular effects. They did observe that it slowed the heart but otherwise thought it an uninteresting drug. In 1941, D Glick noted that cholinesterase metabolized succinylcholine, but again missed succinylcholine’s neuromuscular blocking properties [14].
1912
Arthur Lawen (a colleague of the more famous Trendelenberg), a surgeon in Leipzig, used curare to “soften” the abdominal muscles to facilitate wound closure. He even practiced tracheal intubation and controlled ventilation. However, supplies of curare were unreliable and its potency variable so he abandoned the practice [15]. Some anesthesiologists might find it hard to accept that the first physician to use curare in the operating room was a surgeon.
1928
Francis de Caux, an anesthetist at the North Middlesex Hospital in London, gave curare to patients [16]. Supplies and purity were variable, and he treated only 7 patients:
“When I used Curare in 1928 I used a watery extract of crude curare made by the North Middlesex Hospital Dispensary. It was because there was no method of standardization that we dropped the method because of the inconsistent results obtained’I wrote to two firms Hoffman la Roche and Zimmermans, I believe, who were not very helpful.”
He did not publish his results, and 14 years passed before Griffith and Johnson repeated his experiment in Montreal.
1930s
The use of curare was refined by trial and error until it could be applied safely, but not successfully, in patients with tetanus and other spastic disorders. Interestingly, a group of pathologists (no doubt familiar with the consequences of the failed treatments of tetanus) led by Howard Florey suggested the humane treatment of patients with tetanus by a combination of general anesthesia, paralysis with curare, and artificial ventilation with an iron lung. Although they never tried it, they had predicted what would become standard ICU practice [17].
1935
In 1935, Harold King (1887–1956) extracted tubocurarine from a crude preparation of unknown origin. From the information he gathered, he published an erroneous chemical structure of tubocurarine (Fig. 50.3) [18]. King proposed that curare had quaternary nitrogen groups at each end of the molecule (i.e. it wasbisquaternary). This notion underlay the subsequent development of new muscle relaxants [18]. Discovery of this error in 1970 facilitated development of a new generation of monoquaternary relaxants, the prototype being vecuronium. Although in 1935, large-scale production of tubocurarine was not possible, there followed a string of fortunate circumstances, leading to the first public demonstration of curare in humans.
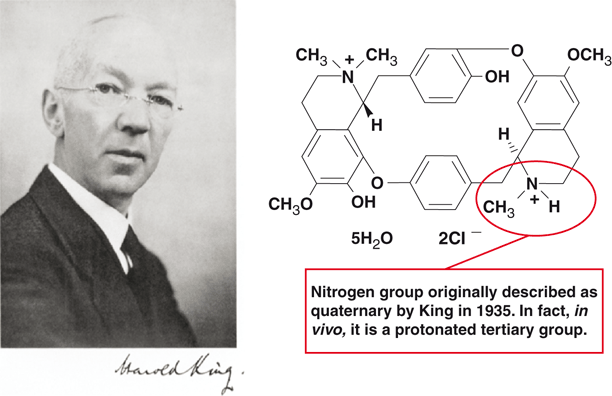
Fig. 50.3
Harold King was the first to isolate and describe the structure of curare. He actually made a minor error and described the group circled as quaternary when in fact it is a protonated tertiary group. This misled scientists for the next three decades to incorrectly think that drugs that had a tertiary group at this site would not be relaxants
1938
Richard Gill solved the problem of inadequate supplies of crude curare. In the course of South American travels, Gill learned how to make crude curare, becoming an honorary witch doctor in the process. When he developed multiple sclerosis he saw the potential for a cure in curare. He brought substantial supplies of curare extract back from Ecuador but the firm Merck that had agreed to process his samples changed their mind and instead pursued investigations into a different plant alkaloid, “erythroidine”.
Gill then offered his supplies to various investigators, including AR McIntyre, Chair of Pharmacology at the University of Nebraska. McIntyre had a research grant from the pharmaceutical company Squibb, and they collaborated to purify the crude extract. A Squibb chemist, Horace Holaday, devised the rabbit head drop test (the name describes the test) to assay the potency of the purified drug [19]. Aliquots of the Intocostrin were administered to the rabbit until the neck muscles no longer kept the head up. Purifying the curare and defining its potency opened the door to therapeutic success.
Success at Last
The psychiatrist Abram Bennet was a colleague of Michael Burman, an orthopedic surgeon who had reported his use of curare in childhood spasticity. Bennet saw a potential for the use of curare to minimize the traumatic consequences of convulsive shock therapy [2,20]. Metrazol-induced convulsions produced a 40–50% incidence of compressive spinal fractures. As reported by Bennet, curare prevented these fractures [20], Such results showed the effectiveness of curare, and probably preserved the practice of convulsive therapy.
The 1940s: It All Comes Together
Because de Caux had not pubished his results, the work had gone unnoticed, and curare was not used in anesthetic practice. Serendipity triggered a change. In 1940, Bennett showed a film of his use of curare to the 91st annual session of the New Jersey Academy of Science. Present was Lewis Wright, Medical Advisor to the pharmaceutical company Squibb. Wright thought curare might be useful in anesthesia. He obtained a preparation of curare, Intocostrin, and donated some to EA Rovenstine at New York University. Rovenstine instructed his resident EM Papper to try Intocostrin in two patients. Both patients became apneic, and Papper had to manually ventilate their lungs overnight. Stuart Cullen at the University of Iowa, also received Intocostrin, used it in dogs, and pronounced “Intocostrin has no place in anesthesia” because the dogs developed bronchospasm. Things did not look good for curare, but this then changed dramatically.
1942 Griffith and Johnson: The Report That Changed Everything
In 1942, a modest paper, brief and to the point, altered forever the practice of anesthesia [21]. With little fanfare, no IRB approval, no oversight, and no previous experience, Harold Griffith and Enid Johnson at the Homeopathic Hospital of Montreal administered Intocostrin to 25 patients anesthetized with cyclopropane. All patients breathed spontaneously throughout surgery, none had their tracheas intubated, and some were challenging (e.g., a 112 kg patient for hemorrhoidectomy and an “…extremely obese woman…” for dilatation and curettage.) Griffith opined: “we have been so much impressed by the dramatic effect produced in every one of our patients that we believe this investigation should be continued.” It is tempting to speculate on what might have followed had any of these patients died from what Beecher might have labeled a “curare death” (see below).
In 1943, two chemists at Squibb purified d-tubocurarine from the bark of Chondodendron Tomentosum. King had extracted the same compound in 1935 but had not known the source of his preparation. The purified d-tubocurarine had 6 times the potency of Intocostrin, and soon replaced it.
1946
In Great Britain, TC Gray and J Halton published an enthusiastic report of the use of both Intocostrin and the purified d-tubocurarine [22]. In 1946 and for sometime thereafter, most patients given curare breathed spontaneously, often without tracheal intubation. It required great skill and vigilance to administer the drug safely, a point made forcibly by Gray and Halton. By 1952, it was appreciated that safe use required control of ventilation [23]. Safety also mandated the postoperative use of doses of up to 5 mg neostigmine to antagonize the residual effect, a use advocated in 1946 [24].
What Does Paralysis Feel Like?
To better understand the patient’s potential perception of curare’s effects, some doughty clinicians submitted to paralysis while awake. In 1943, Helen Barnes underwent laryngoscopy after her colleagues gave her 4 ml of Intocostrin IV while she was awake. Her colleagues passed a laryngoscope and viewed her vocal cords with ease. She then used Intocostrin to facilitate tracheal intubation in 5 patients [25], recognizing that these doses of curare necessitated pharmacologic reversal with neostigmine. In 1946, Frederick Prescott, a pharmacologist at the Westminster hospital in London, submitted to awake paralysis with Intocostrin, a reportedly unpleasant experience during which he remained aware [24].
Despite the reports of Barnes and Prescott, some clinicians believed that curare depressed the central nervous system and could produce anesthesia, and many patients suffered awareness under anesthesia in those early years. “I…was awakened by the most excruciating pain in my tummy. It felt as if my whole insides were being pulled out…” [26].
Scott Smith, a pediatric anesthesiologist in Salt Lake City thought that curare anesthetized babies because they remained immobile during surgery. To test his belief he had colleagues completely paralyze him with curare with support of his respiration by bag and mask [27]. In contrast to what he expected, he perceived touch and pain throughout his paralysis and required tracheal intubation.
Many anesthetists initially thought that the use of muscle relaxants was inherently unsafe, a correct perception for partially paralyzed patients allowed to breathe spontaneously and whose tracheas were not intubated. Some suggested that curare had inherent cardiotoxicity [28]. However in the ten years following its introduction, it became clear that inappropriate use rather than curare caused the problems.
1947
The enthusiastic adoption of curare soon strained supplies, and the need for a synthetic drug became apparent. In response, the Swiss chemist, Daniel Bovet (1907–1992), and his group created gallamine [29]. Gallamine had some clinical success, but Bovet’s work in the next decade with the older drug, succinylcholine, had a much greater impact.
The 1950s: Things Nearly Come Undone
Succinylcholine Appears in 1951
The decade opened propitiously, with the 1951 clinical introduction of succinylcholine. Although succinylcholine was first synthesized in 1906, it fell to Bovet in 1949 to observe its paralyzing action [30]. This observation, and his earlier discovery of gallamine, resulted in his 1957 Nobel Prize for Medicine.
In contrast to Intocostrin, succinylcholine received a warm welcome. Frances Foldes stated “Its use is not accompanied by unwanted side-effects and the incidence of post-operative complications after its use is low”[31]. But first impressions can be deceptive. Subsequently, numerous adverse effects of succinylcholine were identified [6]. One, prolonged block in patients with plasma cholinesterase deficiency, was an early example of the influence of pharmacogenetics, a term coined in 1957 (see Chapter44 on A History of Pharmacogenomics Related to Anesthesiology). The familial nature of prolonged block was soon identified [32], and its relation to atypical cholinesterase demonstrated [33].
Curare and Mortality
In 1954, the “honeymoon” ended. Beecher and Todd reported a six-fold increase in “anesthesia-related” deaths in patients receiving “curare” [28]. To Beecher and Todd, “curare” encompassed d-tubocurarine, Intocostrin, dimethyl-tubocurarine (metubine), gallamine, succinylcholine, and decamethonium: the overall risk of death from anesthesia at that time was 1 in 2100 (far greater than today), but in a patient given curare the risk increased to 1 in 370. No cause was determined, and the increased mortality was not related to the patient’s medical status, experience of the provider, or any other factor that could be identified. Reversal with an anticholinesterase was not reported in the study and presumably was not accomplished. The failure to administer neostigmine almost certainly underlay the lethality of the administration of curare, but in the opinion of the authors, most of the anesthesia-related deaths were from cardiovascular collapse, not respiratory insufficiency. This finding could have ended the clinical use of muscle relaxants, but Beecher and Todd wisely avoided simplistic conclusions. Although the use of relaxants increased anesthesia-related mortality, it did not appear to increase, indeed possibly decreased, overall operative mortality, a point argued forcefully by Abajian and colleagues in a rebuttal paper [34]. Beecher and Todd presciently reasoned that if curare caused an anesthesia-specific problem then the professionalism of anesthesiologists would compel them to investigate, understand, and resolve it.
Mechanisms and Murder in 1956
The depolarizing action of succinylcholine was not immediately appreciated. William Paton delineated relaxants into depolarizing and non-depolarizing drugs in 1956 [35]. Nondepolarizing drugs had a bulky structure (were pachycurares); depolarizing drugs had a long narrow structure (were leptocurares). We are still not sure how relaxants work. Do they enter and plug up ion channels? Do they compete with acetylcholine for receptors on the motor nerve terminal? Do they block the release of acetylcholine? Probably all of the above, and more [36].
In the body, succinylcholine quickly degraded to the naturally occurring compounds choline and succinate. Their natural nature made them nearly undetectable and possibly made succinylcholine the “ideal murder weapon.” Lurid cases of suspected murder by succinylcholine have occurred. In 2007 a Nevada jury convicted critical care nurse, Chaz Higgs, of first degree murder in the death of his wife, Kathy Augustine, a prominent Nevada political figure. The murder weapon was a syringe of succinylcholine [37].
1958
One limitation of curare had been its capacity to cause histamine release and ganglionic blockade’with subsequent low blood pressure and bronchospasm (asthma). One plant species providing raw curare was Strychnos Toxifera. Another alkaloid,alcuronium, extracted from this plant was introduced in 1958. Alcuronium had properties similar to tubocurare but significantly less propensity to produce histamine release and ganglion blockade [38].
The 1960s: Pancuronium, Drug Design, and David Savage
Structure-Function and Steroidal Molecules
This decade saw structure-function analysis applied to drug design. The discovery of a steroidal alkaloid malouetine prompted development of steroid molecules with curare-like effects. In 1935, King had described the structure he thought was required in a neuromuscular blocking drug: two quaternary nitrogen groups separated by a distance (interonium) of 1.4 to 1.5 nm. The interonium distance proved to be more flexible than King predicted, for pancuronium being 1.05 nm and for fazadinium 0.7 nm [39,40]. The general structure had two acetylcholine-like groups separated by an appropriate distance, and had to be bulky (pachycurare) to confer non-depolarizing characteristics. William “Bill” Bowman was a gifted pharmacologist at the University of Strathclyde in Glasgow, Scotland, who had worked with Paton on curare-like drugs. Bowman considered that the rigid steroidal androstane molecule was the perfect skeleton on which to place the quaternary nitrogen groups to create a muscle relaxant drug.
It was these structural “rules” and Bowman’s idea to use the androstane nucleus that led to the development of the aminosteroid relaxant pancuronium by Hewett and Savage [41]. Pancuronium obeyed the rules except for its short interonium distance. This shortness conferred high potency and was long enough to avoid producing ganglionic blockade. Soon after the report of its clinical use in 1967 [42], pancuronium supplanted curare and gallamine for use in patients. Although devoid of ganglionic blocking effects, it had both vagolytic and sympathomimetic activity, but did not produce the hypotension associated with d-tubocurarine, or as much tachycardia as seen with gallamine.
David Savage
The success of pancuronium heralded the emergence of West Scotland as the epicenter for muscle relaxant development for the next 25 years. Leading this effort was David Savage, the principal chemist-scientist working on these compounds at Organon’s research laboratories (Fig. 50.4). He had notable and inventive enthusiasm. He claimed, a bit (but just a bit) tongue in cheek, that using an androstane (steroid) backbone he could create a drug that would work at any receptor. He said that for fun he had made and tested a steroidal appetite suppressant in rats. It was all too good; the rats starved to death! He was also abon viveur. For one particular experiment he needed a small pressure vessel to contain the reaction. The ideal vessel turned out to be a champagne bottle, and of course the contents had to be drunk first.
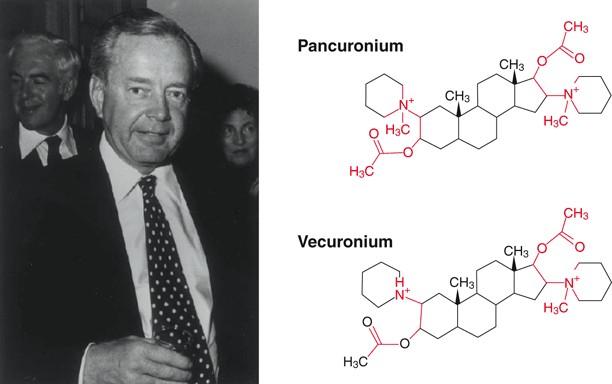
Fig. 50.4
David Savage and his creations, pancuronium and vecuronium. Vecuronium was originally rejected because it was monoquaternary (onered group) as opposed to thebisquaternary pancuronium (twored groups). Sadly, the laboratory where he worked, and where these drugs plus rocuronium and sugammadex were developed has recently closed due to mergers within the pharmaceutical industry
Savage had the then novel insight that scientists and clinicians should work together to develop drugs clinicians needed. So he worked closely with anesthesiologists: Leslie Baird at Glasgow Royal Infirmary, and Ronald Miller in San Francisco. There is a story that one of the brain storming sessions occurred at a local bar. Purely by chance, David Savage and a local anesthesiologist struck up a conversation, and started discussing curare and the neuromuscular junction. In the course of the conversation many novel drug structures, probably including a pancuronium prototype, were written down on paper napkins. So engrossed were the two men that they had to be asked to leave several hours later, as the bartender wanted to close up. Many clinician colleagues of David Savage describe similar “design” sessions, where they would articulate their needs for a drug, and David would draw putative structures. To further scientist/clinician interaction, Savage organized the 1st International Neuromuscular Meeting, bringing clinicians and scientists together in London in 1975.
Like its peers, pancuronium had too long a duration of action because its elimination was primarily via the kidney. Continued research at the Scottish facility solved this problem, leading to the development of two successful drugs, vecuronium and rocuronium, and one ill-fated drug, rapacuronium [43].
Up to the 1970s, clinicians had no practical way of monitoring relaxant effect, except for subjective clinical signs such as abdominal muscle relaxation or adequacy of spontaneous respiration. The need to develop a method of monitoring the residual effects of such powerful drugs was addressed in the next decade.
The 1970s: Monitoring; Ideal Drug; Pharmacokinetics and Pharmacodynamics
Monitoring
In the 1950s and 1960s, clinical acumen and judicious dosing guided the administration of muscle relaxants. The first monitors of neuromuscular function were simple nerve stimulators [44]. They required subcutaneous needles for nerve stimulation, The need to define a control response before relaxant administration made them impractical for normal clinical use. In the 1970s, this led Hassan Ali to develop the train-of-four ratio (TOF; Fig. 50.5) [45]. Ali used the known effect of repeated stimulation (repeated stimulation produces a decreasing contraction in a partly paralyzed muscle) in a clinically useful way [46]. Patients received four stimuli at 2 Hz (0.5 s intervals) and the ratio of the fourth response (T4) to the first (T1) was measured. A material decrease, especially below 0.7, indicated residual paralysis.

Fig. 50.5
Significant figures in the modern history of neuromuscular function. Hassan Ali in Boston developed the concept of the train-of-four ratio to quantify block. Jørgen Viby-Mogensen in Copenhagen took this work further and developed the techniques of post-tetanic count to quantify profound block, and double burst stimulation to facilitate clinical detection of low levels of residual block. John Savarese, originally in Boston and later in New York developed mivacurium and has had a career-long pursuit of a type A fast-onset short acting non-depolarizer muscle relaxant. Finally, Anton “Ton” Bom, a native of Holland, working in Scotland, changed the whole concept of relaxant reversal with the discovery and development of sugammadex
This useful ratio (Fig. 50.6) correlated with other indices of muscle function, including the absolute decrease in strength of the muscle contraction, the decrease in response to tetanic stimulation, and deteriorating pulmonary function [47,48]. Because it provided its own internal control, it did not require a baseline measurement. The response could be either mechanical force, or electromyography. It also discriminated between succinylcholine-induced block (minimal fade) and depolarizing (marked fade) block. TOF assessment persists as the mainstay of clinical monitoring.
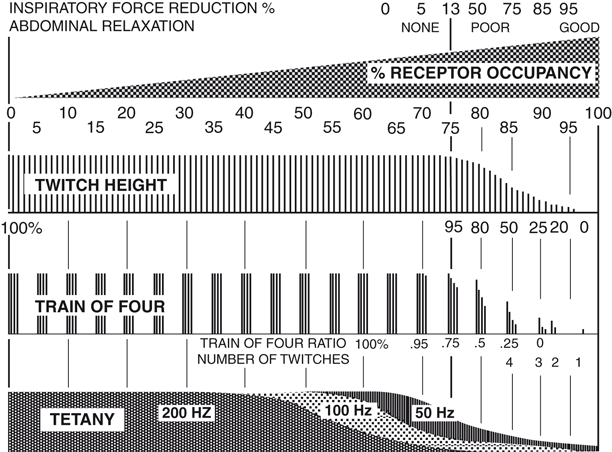
Fig. 50.6
The three most common stimulation modalities in neuromuscular monitoring are single twitch height, train-of-four and tetanus at 50 Hz. They have a fairly reproducible relationship to one another and to the fraction of acetylcholine receptors that need to be blocked (occupied) by the muscle relaxant for a given effect. (From an online lecture Thompson C: Monitoring the Neuromuscular Junction, University of Sydney, 2010, with permission.)
The Ideal Drug
In the 1970s, clinicians could choose a short-acting drug (succinylcholine) or longer acting (nondepolarizing) drugs. All had side effects. This unsatisfactory situation led to the concept of the ideal drug by Karis and Gissen in 1971 [49]. Their criteria have stood the test of time and are still quoted today (Table 50.2), with the notable exception that a demand for fast onset was absent.
Table 50.2
Definition of the “Ideal Drug” Karis and Gissen
Criterion | Comment |
|---|---|
1. Competitive non-depolarizing | Avoided complications of depolarizing action |
2. Recovery not organ-dependent | Fulfilled by atracurium and mivacurium |
3. Breakdown products inactive | Fulfilled by rocuronium and cisatracurium |
4. Relatively short duration | Fulfilled by intermediate acting drugs |
5.Highly specific | Delayed development of drugs with fast onset |
6. Reversible by anticholinesterase | Did not foresee alternatives |
Savarese and Kitz elaborated on these concepts in 1975 [50], adding their belief that one drug could not be ideal for all clinical situations. They defined three categories of ideal drug, A, B and C. Type A was a non-depolarizing succinylcholine (rapid onset, rapid offset). Type B had a duration of action approximately half that of curare or pancuronium, and soon became a reality with the introduction of vecuronium and atracurium. Type C was long acting and without adverse cardiovascular effects, anticipating pipecuronium and doxacurium which arrived approximately 15 years later. John Savarese has crusaded for a Type A drug, and, as we shall see, his efforts may be close to fruition.
Defining the direction for new drugs pressured the pharmaceutical industry to synthesize products that would meet the challenges implied by the definitions. As noted earlier, Savage facilitated the dialogue between clinicians and industry by organizing the 1st Neuromuscular Meeting in London in 1975, just when the concept of the “ideal relaxant” was described. Few at the meeting appreciated the impending “revolution” in neuromuscular pharmacology’the era of the intermediate-acting relaxant was about to dawn.
Pharmacokinetics and Pharmacodynamics
Until the 1970s, there was but a rudimentary understanding of drug actions, and of the interaction of biodisposition (pharmacokinetics) and effect (pharmacodynamics). Developments at the University of California San Francisco (UCSF) changed this (Fig. 50.7). Leslie Benet developed and refined the science of pharmacokinetics and pharmacodynamics, creating the concept of drug “Clearance.” Concurrently, Lewis Sheiner pursued the science of drug concentration-effect relationships. And a young clinician/scientist Ronald Miller, freshly back from Vietnam, had a mind full of questions relating to the clinical behavior of muscle relaxants and their antagonists. These circumstances fortuitously combined a clinician with important questions and scientists with the talents to develop the tools to address those questions. The breakthrough was to measure both the effect of a drug and the concentrations associated with the effect, and to make inferences as to how underlying conditions (e.g., renal or hepatic failure) influenced these. The first paper applying these techniques focused on the impact of renal failure on the pharmacokinetics of tubocurare [51].

Fig. 50.7
The University of California San Francisco threesome that significantly advanced the scientific understanding of the pharmacokinetics and pharmacodynamics of muscle relaxants in the 1970s. Leslie Benet created the pharmacokinetic concept of “Clearance.” Concurrently, Lewis Sheiner pursued the science of drug concentration-effect relationships, developing sophisticated modeling tools. Finally, a young clinician/scientist Ronald Miller, freshly back from Vietnam, had many questions relating to the clinical behavior of muscle relaxants and their antagonists. Good fortune brought together a clinician with important questions and scientists with the talents to develop the tools to address those questions
A problem remained: the concurrent measurement of the concentration of a drug at its site of action (e.g., the neuromuscular junction) and the effect of that concentration. The researcher couldn’t sample the neuromuscular junction. Surrogate measures (e.g., the concentration in blood) had to serve instead. That was fine (sort of) if there were equilibrium between the blood and the neuromuscular junction, but how could one be sure? Sheiner and Stanski published an important paper in 1979 that provided the “missing link” between concentration and effect [52]. The authors in this paper recognized that initially the concentration at the neuromuscular junction (and the effect) lagged behind what the anesthetist gave and was in the blood. During recovery, the opposite was true. The paper demonstrated how one could calculate the concentration and effect at the neuromuscular junction by knowing the effect during input and output. This model has broad applicability to any situation where the drug concentration is measured in one place and its effect takes place at another.
Related posts:
 History Reflected in the Evolving Approaches to Anesthesia for a Patient Undergoing Cholecystectomy
A History of Intravenous Anesthesia
History Reflected in the Evolving Approaches to Anesthesia for a Patient Undergoing Cholecystectomy
A History of Intravenous Anesthesia
 Major Anesthesia-Related Events in the 2000s and Beyond
Major Anesthesia-Related Events in the 2000s and Beyond
 1846–1860: Following the Discovery of Anesthesia
1846–1860: Following the Discovery of Anesthesia
 Major Anesthetic Themes in the 1960s
Major Anesthetic Themes in the 1960s
 A History of Intensive Care Medicine
A History of Intensive Care Medicine
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


