Perioperative blood component therapy accounts for at least 23% of transfusions.
 Blood must not only be maintained as a fluid in normal circulation, but also be capable of forming a solid clot to stanch leaks in the vascular wall—and then dismantling the clot when the need has passed.
Blood must not only be maintained as a fluid in normal circulation, but also be capable of forming a solid clot to stanch leaks in the vascular wall—and then dismantling the clot when the need has passed.
 Clotting factors in the plasma are activated at sites of endothelial injury and assemble in enzymatic complexes to activate thrombin.
Clotting factors in the plasma are activated at sites of endothelial injury and assemble in enzymatic complexes to activate thrombin.
 Fibrin clots must be broken down after their job is done, and fibrinolysis is a complex process with checks and balances.
Fibrin clots must be broken down after their job is done, and fibrinolysis is a complex process with checks and balances.
 The first screening test for hemostatic problems should always be the patient’s medical history.
The first screening test for hemostatic problems should always be the patient’s medical history.
 Platelet aggregation is the most detailed overall platelet function test.
Platelet aggregation is the most detailed overall platelet function test.
 A general oversight of plasma clotting factor activity is obtained by the prothrombin time (PT) for the intrinsic (tissue) pathway and the activated partial thromboplastin time (aPTT) for the extrinsic (contact) pathway.
A general oversight of plasma clotting factor activity is obtained by the prothrombin time (PT) for the intrinsic (tissue) pathway and the activated partial thromboplastin time (aPTT) for the extrinsic (contact) pathway.
 Disseminated intravascular coagulopathy (DIC) describes unchecked coagulation initiated by pathologic systemic activation of the intrinsic clotting pathway.
Disseminated intravascular coagulopathy (DIC) describes unchecked coagulation initiated by pathologic systemic activation of the intrinsic clotting pathway.
 The risk for venous thromboembolism is increased by intercurrent factors such as physical inactivity or immobilization, malignancy, oral contraceptives, estrogen therapy, and pregnancy.
The risk for venous thromboembolism is increased by intercurrent factors such as physical inactivity or immobilization, malignancy, oral contraceptives, estrogen therapy, and pregnancy.
 Most anticoagulant therapies need ongoing or selective testing for assessment of therapeutic effect.
Most anticoagulant therapies need ongoing or selective testing for assessment of therapeutic effect.
 Leukoreduction to remove WBCs from RBCs and platelets reduces the risk of HLA alloimmunization, febrile nonhemolytic transfusion reactions, and CMV transmission in patients who require these precautions.
Leukoreduction to remove WBCs from RBCs and platelets reduces the risk of HLA alloimmunization, febrile nonhemolytic transfusion reactions, and CMV transmission in patients who require these precautions.
 Plasma derivatives are proteins processed from plasma for therapeutic infusions.
Plasma derivatives are proteins processed from plasma for therapeutic infusions.
 Techniques have been developed to kill microbial pathogens in blood components.
Techniques have been developed to kill microbial pathogens in blood components.
 Many years of effort have gone into the search for an oxygen-carrying substitute for RBCs.
Many years of effort have gone into the search for an oxygen-carrying substitute for RBCs.
 Routine RBC compatibility testing includes ABO and RhD typing, an antibody screen for IgG non-ABO RBC antibodies, and an RBC cross-match.
Routine RBC compatibility testing includes ABO and RhD typing, an antibody screen for IgG non-ABO RBC antibodies, and an RBC cross-match.
 Over the past decade, transfusion practices for medical and surgical patients shifted from a liberal strategy to more restrictive management with lower thresholds and careful consideration of the balance between transfusion risks and the physiologic consequences of anemia.
Over the past decade, transfusion practices for medical and surgical patients shifted from a liberal strategy to more restrictive management with lower thresholds and careful consideration of the balance between transfusion risks and the physiologic consequences of anemia.
 Oxygen delivery to the tissues (DO2) is dependent on cardiac output (CO), regional blood flow, and oxygen-carrying capacity also known as the oxygen content (CaO2) of blood.
Oxygen delivery to the tissues (DO2) is dependent on cardiac output (CO), regional blood flow, and oxygen-carrying capacity also known as the oxygen content (CaO2) of blood.
 Numerous recommendations provide guidance for the transfusion management of thrombocytopenia and acquired or inherited platelet disorders.
Numerous recommendations provide guidance for the transfusion management of thrombocytopenia and acquired or inherited platelet disorders.
 Cryoprecipitate is created by a controlled thaw of frozen plasma which allows for precipitation of large molecules.
Cryoprecipitate is created by a controlled thaw of frozen plasma which allows for precipitation of large molecules.
 Over the past few decades, the risk benefit ratio of blood product transfusion has been the subject of several studies and review articles.
Over the past few decades, the risk benefit ratio of blood product transfusion has been the subject of several studies and review articles.
 Given the extensive use of more sensitive methods for screening and controlling the infectious risks of blood product transfusion, noninfectious complications have emerged as the major source of transfusion-related morbidity and mortality.
Given the extensive use of more sensitive methods for screening and controlling the infectious risks of blood product transfusion, noninfectious complications have emerged as the major source of transfusion-related morbidity and mortality.
 Transfusion-related acute lung injury (TRALI) is a clinical diagnosis that can be clouded by confounding comorbidities or patient acuity; therefore, TRALI tends to be underreported in the literature and is extremely difficult if not impossible to study with randomized prospective clinical trials.
Transfusion-related acute lung injury (TRALI) is a clinical diagnosis that can be clouded by confounding comorbidities or patient acuity; therefore, TRALI tends to be underreported in the literature and is extremely difficult if not impossible to study with randomized prospective clinical trials.
 Acute normovolemic hemodilution is the process of extracting multiple units of blood immediately before surgical incision while maintaining euvolemia with crystalloids or colloid supplementation.
Acute normovolemic hemodilution is the process of extracting multiple units of blood immediately before surgical incision while maintaining euvolemia with crystalloids or colloid supplementation.
 Over the past decade, RBC salvage techniques have improved drastically and now offer an efficient, cost-effective, and safe method for perioperative blood conservation.
Over the past decade, RBC salvage techniques have improved drastically and now offer an efficient, cost-effective, and safe method for perioperative blood conservation.
 Disorders of hemostasis can be classified as those that cause a propensity for hemorrhage and those that facilitate inappropriate thrombosis.
Disorders of hemostasis can be classified as those that cause a propensity for hemorrhage and those that facilitate inappropriate thrombosis.
 Symptomatically disorders of primary hemostasis often present with superficial signs of bleeding on the skin or mucosa.
Symptomatically disorders of primary hemostasis often present with superficial signs of bleeding on the skin or mucosa.
 Von Willebrand disease (vWD) is the most common hereditary bleeding disorder with a prevalence of approximately 1% in the general population.
Von Willebrand disease (vWD) is the most common hereditary bleeding disorder with a prevalence of approximately 1% in the general population.
 Hemophilia is a genetic disease that results from deficiencies or dysfunction of specific clotting factors.
Hemophilia is a genetic disease that results from deficiencies or dysfunction of specific clotting factors.
 Antiplatelet therapy is indicated for patients at risk of cerebral vascular accident, myocardial infarction, or other vascular thrombosis complications.
Antiplatelet therapy is indicated for patients at risk of cerebral vascular accident, myocardial infarction, or other vascular thrombosis complications.
 Heparin-induced thrombocytopenia (HIT) is a clinical disorder that develops after extended use of heparin therapy. It occurs in approximately 1% to 5% of patients receiving heparin and is associated with morbidity from thromboembolic complications.
Heparin-induced thrombocytopenia (HIT) is a clinical disorder that develops after extended use of heparin therapy. It occurs in approximately 1% to 5% of patients receiving heparin and is associated with morbidity from thromboembolic complications.
 Recombinant activated factor VII (rFVIIa) is now indicated for the treatment of acquired hemophilia and factor VII deficiency.
Recombinant activated factor VII (rFVIIa) is now indicated for the treatment of acquired hemophilia and factor VII deficiency.
 Prothrombin Complex Concentrates are now the drug of choice for emergent reversal of oral anticoagulants in place of rFVIIa and fresh frozen plasma.
Prothrombin Complex Concentrates are now the drug of choice for emergent reversal of oral anticoagulants in place of rFVIIa and fresh frozen plasma.
 Antifibrinolytic agents have been used to prevent and treat surgical blood loss for several decades.
Antifibrinolytic agents have been used to prevent and treat surgical blood loss for several decades.
Multimedia
 Cerebral Aneurysm Coiling
Cerebral Aneurysm Coiling
 Formation and Lysis of Fibrin
Formation and Lysis of Fibrin
INTRODUCTION
Over 23 million blood products were transfused throughout the United States during the year 2008 alone. This included over 15 million units of red blood cells (RBCs), about 2 million packs of platelets, over 4.4 million units of plasma, and approximately 1.1 million doses of cryoprecipitate.  Perioperative blood component therapy accounts for at least 23% of these transfusions.1 Consequently, it is imperative for the anesthesia provider to understand the treatment benefits, the rare and common adverse effects, and the specific therapeutic details of blood product preparation and delivery in order to best manage their patients.
Perioperative blood component therapy accounts for at least 23% of these transfusions.1 Consequently, it is imperative for the anesthesia provider to understand the treatment benefits, the rare and common adverse effects, and the specific therapeutic details of blood product preparation and delivery in order to best manage their patients.
This chapter begins with a review of primary and secondary hemostasis, fibrinolysis, and regulation of the coagulation pathway. We continue with a description of the most common coagulation profile tests followed by the method for blood product collection and storage. The therapeutic indications and risks associated with blood component therapy are discussed at length. The chapter also includes extensive clinical sections discussing congenital and acquired deficiencies in hemostasis and coagulation, as well as an up-to-date presentation of available pharmacologic treatment medications to maintain a balanced hemostatic mechanism.
HEMOSTASIS AND COAGULATION
Primary Hemostasis
 Blood must not only be maintained as a fluid in normal circulation, but also be capable of forming a solid clot to staunch leaks in the vascular wall—and then dismantling the clot when the need has passed. This delicate equilibrium between anticoagulation and coagulation is maintained by a complex system of counterbalanced blood proteins and cells (platelets). Many congenital and acquired disorders can push the system toward either bleeding or thrombosis. The patient care team has a number of tests to evaluate the system and many therapeutic modalities to correct these imbalances.
Blood must not only be maintained as a fluid in normal circulation, but also be capable of forming a solid clot to staunch leaks in the vascular wall—and then dismantling the clot when the need has passed. This delicate equilibrium between anticoagulation and coagulation is maintained by a complex system of counterbalanced blood proteins and cells (platelets). Many congenital and acquired disorders can push the system toward either bleeding or thrombosis. The patient care team has a number of tests to evaluate the system and many therapeutic modalities to correct these imbalances.
Platelets adhere to sites of endothelial disruption, undergo activation to recruit more platelets and amplify the platelet response, and then cross-link with fibrin, the end product of the plasma clotting factor cascade, to form a platelet plug. Primary hemostasis (Fig. 16-1) describes the initiation of the platelet plug and clotting mechanism.
FIGURE 16-1. Overview of platelet pathways for adherence, activation, stabilization, and physiologic inhibition. Thin arrows, signaling pathways; thick arrows, ligand binding; curved arrows, catalysis; clear arrows, secretion; slashed circles, inhibitory signaling pathways; round circles, antiplatelet drug targets; pentagons, targets of antiplatelet drugs in development. (A) Adherence: vWF, von Willebrand factor. Glycoproteins Ib/IX, Ia/IIa, and VI. (B) Activation: Agonists: TxA2, thromboxane A2; ADP, adenosine diphosphate. Receptors: GPCR, G-protein–coupled receptor; TP, thromboxane prostanoid; PAR, protease-activated receptor. Intermediaries: PLC, phospholipase C; IP3, inositol-1,4,5-triphosphate; DAG, diacylglycerol; PKC, protein kinase C; Ca2+, calcium; PLA2, phospholipase A2; AA, arachidonic acid; COX, cyclooxygenase. (C) Stabilization: Glycoprotein IIb/IIIa. (D) Inhibition: NO, nitric oxide; PGI2, prostaglandin I2 (prostacyclin); IP, PGI2 receptor; AC, adenylate cyclase; cAMP, cyclic adenosine monophosphate; PDE, phosphodiesterase; cGMP, cyclic guanosine monophosphate; PKA, protein kinase A; PKG, protein kinase G. Targets of antiplatelet drugs. COX-1: Aspirin and nonsteroidal antiinflammatory drugs (NSAIDs). P2Y12: Clopidogrel, prasugrel, ticlopidine, and, in clinical trials, cangrelor, elinogrel, and ticagrelor. cAMP PDE: Dipyridamole and cilostazol. IIb/IIIa: Abciximab, eptifibatide, and tirofiban. Other drugs in clinical trials—TP: Terutroban; PAR-1: E5555, SCH 530348; vWF: AJW200, ARC1779.
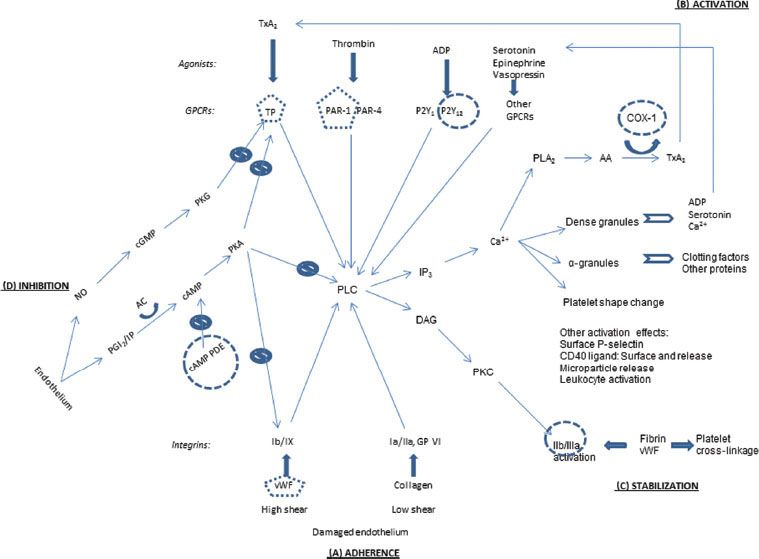
Adherence
When the endothelial lining is disrupted to expose the underlying matrix, platelets attach to collagen via surface integrin receptors—glycoproteins (GP) Ia/IIa and GP VI (Fig. 16-1A). Collagen adherence is favored in low-shear conditions such as venous circulation.2 In high-shear arterial blood flow, von Willebrand factor (vWF) from endothelial cells and from preexisting clot binds to integrin Ib/IX, the other major adherence anchor.3 In capillary blood flow, platelets are pushed to the periphery by RBCs, so anemia lessens platelet contact and reduces platelet function.2
Activation
Platelet activation can be mediated by numerous signaling pathways from the platelet surface (Fig. 16-1B). In “outside-in” signaling, a central target is phospholipase C (PLC). The above adherence integrins trigger pathways to activate PLC.2 Another set of surface receptors, G-protein–coupled receptors (GPCRs), are activated by an array of corresponding agonists, including thrombin from the factor clotting cascade, adenosine diphosphate (ADP), thromboxane A2 (TxA2), serotonin, epinephrine, and vasopressin. Each of these agonist–GPCR pairings set off activation pathways for PLC as well.4
Activated PLC leads to several structural changes in the platelets. Via inositol-1,4,5-triphosphate (IP3), calcium (Ca2+) is released from storage tubules. Calcium ions catalyze release of dense granules and α-granules at the platelet surface. These granules contain ADP, serotonin, and more Ca2+, all of which can activate more platelets.2 α-granules contain numerous proteins, including factor V, fibrinogen, and platelet factor 4 (PF-4), which promotes clotting by binding and neutralizing heparin-like compounds and heparin.2 (This heparin–PF-4 complex is the target antigen for the antibodies causing heparin-induced thrombocytopenia [HIT], discussed in depth later in this chapter.) Calcium also facilitates rearrangement of the platelet microskeleton to change the platelet shape from round and discoid to flat and spiky. Furthermore, the Ca2+ helps activate phospholipase A2 (PLA2), which releases arachidonic acid (AA) from the platelet membrane. AA, as catalyzed by cyclooxygenase-1 (COX-1), is modified to TxA2, which can then activate more platelets.3 Activated platelets also have surface P-selectin and surface-bound and released CD40 ligand, and they release circulating microparticles and attract and activate leukocytes; these features further contribute to hemostasis and also play a role in inflammation.2
Stabilization
The activated PLC initiates “inside-out” signaling of GP IIb/IIIa via diacylglycerol (DAG) and protein kinase C (Fig. 16-1C). This changes the shape of GP IIb/IIIa, which permits it to better bind fibrin and vWF. These proteins can bridge to other activated platelets.3 The fibrin binding can also enmesh the platelets, contributing to the formation of the platelet plug in the convergence of the platelet and clotting factor systems.
Inhibition
To maintain hemostatic balance, platelets are naturally inhibited in their endothelial environment. Endothelial cells secrete prostaglandin I2 (PGI2), which binds to a surface receptor to signal increased cyclic adenosine monophosphate (cAMP). Elevated cAMP activates protein kinase A (PKA), a multisite inhibitor of vWF adherence, TxA2 activation, and PLC internal signaling. However, cAMP is metabolized by cAMP phosphodiesterase (PDE). Endothelial cells also secrete nitric oxide (NO), which at high levels initiates a signaling pathway leading to inhibition of the TxA2 receptor.4
Mechanisms of Antiplatelet Medications
Figure 16-1 shows the sites of action for antiplatelet medications in use or in development. No drugs are available to counteract the first step, platelet adherence (Fig. 16-1A), although at least two compounds are under development to affect vWF’s binding to GP Ib/IX. Aspirin and nonsteroidal antiinflammatory agents dampen the secretion of TxA2 by inhibiting COX-1, the enzyme which converts AA into TxA2 (Fig. 16-1B). Another agonist, ADP, has its P2Y12 receptor blocked by clopidogrel and its analogues. Drugs are in development to block surface receptors for TxA2 and thrombin. The formation and stabilization of the platelet plug is blocked by abciximab, eptifibatide, and tirofiban, which act at GP IIb/IIIa (Fig. 16-1C). Finally, the major inhibitory pathway mediated by endothelial PGI2 is upregulated by dipyridamole and cilostazol (Fig. 16-1D). These medications are discussed further later in this chapter.
Secondary Hemostasis
 Clotting factors in the plasma are activated at sites of endothelial injury and assemble in enzymatic complexes to activate thrombin. This initiates secondary hemostasis. Thrombin then amplifies production of itself by activating other more efficient enzymes which propagate a thrombin burst. Thrombin also converts fibrinogen to fibrin, which cross-links with activated platelets to form the platelet plug. Each of the three enzymatic complexes in the clotting process consists of four parts: An enzyme in the serine protease family, a cofactor, a plasma membrane phospholipid surface such as the platelet, and calcium ion (Ca2+). The proteases convert other clotting factors from their inactive circulating configuration to an active form (termed [factor number]a).5
Clotting factors in the plasma are activated at sites of endothelial injury and assemble in enzymatic complexes to activate thrombin. This initiates secondary hemostasis. Thrombin then amplifies production of itself by activating other more efficient enzymes which propagate a thrombin burst. Thrombin also converts fibrinogen to fibrin, which cross-links with activated platelets to form the platelet plug. Each of the three enzymatic complexes in the clotting process consists of four parts: An enzyme in the serine protease family, a cofactor, a plasma membrane phospholipid surface such as the platelet, and calcium ion (Ca2+). The proteases convert other clotting factors from their inactive circulating configuration to an active form (termed [factor number]a).5
The extrinsic pathway (Fig. 16-2A–E). The process begins when endothelial disruption exposes tissue factor (TF) on underlying cell membranes (A), extrinsic to the circulation—hence the term “extrinsic pathway.” TF binds both VII and VIIa, which circulates at low levels, and is a cofactor for the activation of factor VII (B). VIIa enzyme, TF cofactor, cell membrane phospholipid, and Ca2+ form the first complex, a low-efficiency extrinsic-pathway “tenase” which activates factor X and factor IX (C). Then Xa enzyme, its cofactor Va (derived in large part from factor V released from activated platelet α-granules), phospholipid, and Ca2+ assemble to form the second complex, a “prothrombinase,” which converts prothrombin (II) to thrombin (IIa) (E).5
FIGURE 16-2. Summary of secondary hemostasis and the intrinsic, extrinsic, and common coagulation pathways. See text for details. TF, tissue factor; TFPI, tissue factor pathway inhibitor; vWF-VIII, circulating factor VIII bound to von Willebrand factor. (Reproduced with permission from: John C. Drummond, MD, and Charise T. Petrovitch, MD.)
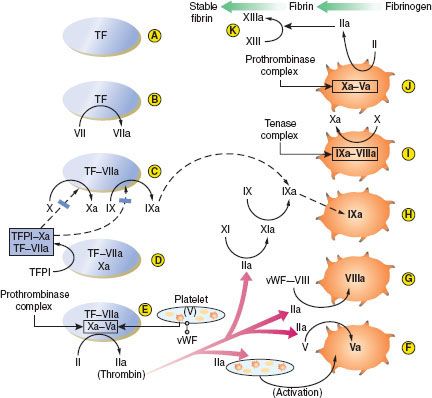
The intrinsic pathway (Fig. 16-2F–J). Thrombin has several central functions. It activates platelets via surface receptors PAR-1 and PAR-4 (see Primary Hemostasis), cleaves more V to Va (F), and initiates the “intrinsic” (intravascular) coagulation pathway by cleaving factor XI to XIa (G). XIa cleaves more IX to IXa (H). Thrombin also activates VIII to VIIIa (G). (VIII is carried and stabilized in the plasma by vWF until needed, and vWF deficiency results in low plasma VIII levels also.) The third complex is then formed: IXa enzyme, VIIIa cofactor, phospholipid, and Ca2+. This is a high-efficiency intrinsic-pathway “tenase” (I) which provides many times more Xa for more prothrombinase complex (J). Ultimately, thrombin cleaves fibrinogen to fibrin monomers, which then polymerize extensively. Fibrin polymers are cross-linked by factor XIIIa (also activated by thrombin) to form the stable fibrin clot (K). Fibrin also cross-links activated platelets by their GP IIb/IIIa receptors to enmesh platelets and fibrin in the platelet plug (see Primary Hemostasis).5
All of these clotting factors are primarily produced in the liver, except for VIII, which is also released by endothelial cells and is well maintained in liver disease. The plasma half-life of most clotting factors is around 1.5 to 3 days, but those of the initiating factor VII (6 hours) and the cofactors V and VIII (8 to 12 hours) are much shorter. Four critical enzyme factors—VII in the extrinsic tenase, IX in the intrinsic tenase, X in the prothrombinase, and prothrombin (II)—must be carboxylated at multiple glutamic acid residues after translation, in order to interact with phospholipid and Ca2+. Vitamin K in its reduced form is the cofactor for the glutamyl-carboxylase enzyme, and thus these factors are vitamin K–dependent.5
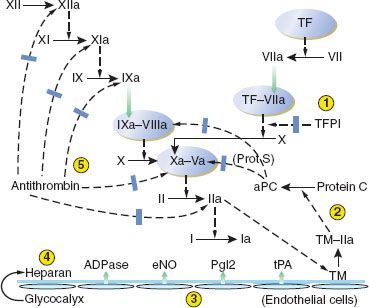
Inhibition of Clotting Factors
The clotting pathways have three main regulatory inhibitors: (1) TF pathway inhibitor (TFPI), (2) antithrombin-III (AT-III), and (3) protein C and its activation by the “protein C-ase” complex (Fig. 16-3).1–5 (1) TFPI inhibits the external tenase complex by binding to the VIIa protease and to its Xa product. TFPI is produced in endothelial cells, from which its release is stimulated by heparin. Heparin in turn binds to and raises the inhibitory efficiency of TFPI. (2) AT-III is a serine protease inhibitor, or serpin. Serpins disrupt the active sites and increase the clearance of their target proteases. AT-III inhibits proteases in all clotting pathways: VIIa in intrinsic tenase, Xa in prothrombinase, XIa and IXa in the intrinsic tenase pathway, and thrombin. AT-III’s inhibitory function is greatly increased when bound to heparin. (3) Protein C-ase is an enzymatic complex with the same structure as the coagulation complexes above: An enzyme, thrombin, its cofactor thrombomodulin, phospholipid, and Ca2+. Thrombomodulin is expressed on endothelial cell membranes. In the protein C-ase complex, thrombin cleaves and activates protein C. Activated protein C (APC) brakes clotting by cleaving VIIIa and Va, the cofactors for the external tenase and the prothrombinase complexes. Protein C has a short half-life of 6 hours. Protein S is thought to be a cofactor for protein C; both are vitamin K–dependent.5
FIGURE 16-3. Depiction of the antithrombotic regulation of hemostasis. Five mechanisms serve to prevent unrestrained coagulation. (1) Tissue factor pathway inhibitor (TFPI) inhibits the initial activation of factor X by the extrinsic pathway. (2) A complex of thrombomodulin (TM) and thrombin (IIa) activates protein C, which, with protein S (Prot S) as a cofactor, inhibits activated factors V and VIII. (3) Intact vascular endothelium releases several substances that have a platelet-inhibiting or clot-lysing effect, including nitric oxide (eNO), prostacyclin (PgI2), adenosine diphosphatase (ADPase), and tissue plasminogen activator (tPA). (4) In addition to TM, other coagulation-inhibiting substances including heparan sulfate and dermatan sulfate (latter not shown) are present in the intact glycocalyx. (5) Antithrombin-III binds, and thereby inhibits, several activated clotting factors (XIIa, XIa, IXa, Xa, and IIa). TF, tissue factor; aPC, activated protein C. (Reproduced with permission from: John C. Drummond, MD, and Charise T. Petrovitch, MD.)
Fibrinolysis
 Fibrin clots must be broken down after their job is done, and fibrinolysis is also a complex process with checks and balances.
Fibrin clots must be broken down after their job is done, and fibrinolysis is also a complex process with checks and balances.  In the end, plasminogen is activated to plasmin, which breaks down fibrin polymers (Fig. 16-4). Plasminogen activation has a minor and a major pathway. In the minor pathway, IXa, XIIa, and kallikrein can each activate plasminogen. The latter two factors are part of the “contact factors” which initiate in vitro clotting in the activated partial thromboplastin (activated partial thromboplastin time [aPTT]) test, but are not thought to take part in clotting in vivo. The minor pathway is thought to account for a small fraction of fibrinolysis.
In the end, plasminogen is activated to plasmin, which breaks down fibrin polymers (Fig. 16-4). Plasminogen activation has a minor and a major pathway. In the minor pathway, IXa, XIIa, and kallikrein can each activate plasminogen. The latter two factors are part of the “contact factors” which initiate in vitro clotting in the activated partial thromboplastin (activated partial thromboplastin time [aPTT]) test, but are not thought to take part in clotting in vivo. The minor pathway is thought to account for a small fraction of fibrinolysis.
FIGURE 16-4. The mechanism and regulation of fibrinolysis. Fibrin is formed from fibrinogen by the action of thrombin (FIIa). Thrombin also converts factor XIII (FXIII) to activated factor XIII (FXIIIa), which in turn stabilizes the evolving fibrin clot by cross-linkage. Circulating plasminogen binds to fibrin and is converted to plasmin by tissue plasminogen activator (tPA) released from normal endothelium in areas remote from sites of vascular injury. Plasmin digests fibrin to its various degradation products (FDPs). The action of tPA can be inhibited by plasminogen activator inhibitor (PAI-1) released by endothelium and platelets. The action of plasmin is also inhibited by thrombin-activated fibrinolysis inhibitor (TAFI). (Reproduced with permission from: John C. Drummond, MD, and Charise T. Petrovitch, MD.)
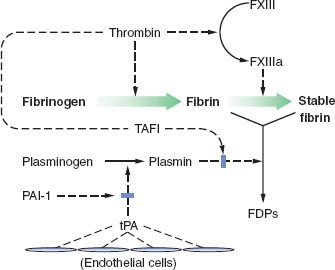
The major activator of plasminogen in the blood is tissue plasminogen activator (tPA), which is secreted from endothelial cells. When associated with cross-linked fibrin, tPA becomes much more efficient. Once some plasmin is formed, it cleaves tPA to a more active form. tPA also directly cleaves fibrin polymers. In tissues, urokinase plasminogen activator is important in fibrinolysis. Urokinase is secreted from the endothelial cells and the kidney. Plasmin also activates urokinase to a more active form.
Inhibition of Fibrinolysis
Plasminogen activation inhibitor-1 (PAI-1) is a serpin which  binds to tPA and urokinase and accelerates their clearance from plasma (Fig. 16-4). Activated platelets release PAI-1 from α-granules. Thrombin-activated fibrinolysis inhibitor (TAFI) is activated by the thrombin–thrombomodulin protein C-ase complex. TAFI cleaves fibrin and fibrin polymers in a fashion which inhibits the action of tPA, and TAFI also inhibits the action of plasmin on fibrin. α2-antiplasmin binds to plasmin and blocks its action, although this also slows the metabolism of plasmin.
binds to tPA and urokinase and accelerates their clearance from plasma (Fig. 16-4). Activated platelets release PAI-1 from α-granules. Thrombin-activated fibrinolysis inhibitor (TAFI) is activated by the thrombin–thrombomodulin protein C-ase complex. TAFI cleaves fibrin and fibrin polymers in a fashion which inhibits the action of tPA, and TAFI also inhibits the action of plasmin on fibrin. α2-antiplasmin binds to plasmin and blocks its action, although this also slows the metabolism of plasmin.
LABORATORY EVALUATION OF HEMOSTASIS
 The first screening test for hemostatic problems should always be the patient’s medical history.6 The nature of any abnormal bleeding is helpful; dermal or mucosal bleeding may suggest platelet dysfunction, whereas hemarthroses or soft tissue bleeding suggests factor deficiencies. Besides any direct past history of bleeding, thrombosis, or laboratory abnormalities, the patient’s experience with hemostatic challenges such as surgery, dental procedures, and menstruation may help rule out clinical problems or suggest a lifelong congenital or more recent acquired disorder. The family history is helpful in diagnosing a congenital problem and the possible pattern of inheritance. Anticoagulants and antiplatelet medications, including over-the-counter drugs, should always be reviewed before ordering laboratory analysis.
The first screening test for hemostatic problems should always be the patient’s medical history.6 The nature of any abnormal bleeding is helpful; dermal or mucosal bleeding may suggest platelet dysfunction, whereas hemarthroses or soft tissue bleeding suggests factor deficiencies. Besides any direct past history of bleeding, thrombosis, or laboratory abnormalities, the patient’s experience with hemostatic challenges such as surgery, dental procedures, and menstruation may help rule out clinical problems or suggest a lifelong congenital or more recent acquired disorder. The family history is helpful in diagnosing a congenital problem and the possible pattern of inheritance. Anticoagulants and antiplatelet medications, including over-the-counter drugs, should always be reviewed before ordering laboratory analysis.
Laboratory Evaluation of Primary Hemostasis
The normal automated platelet count in adults is approximately 150,000 to 400,000/μL. The peripheral blood smear should be examined in specimens with abnormal platelet counts or size. Microscopic review may reveal clotted specimens, artifactual platelet clumping in vitro, or abnormal platelet morphology. Large platelets are seen in some congenital disorders. One of the first platelet function tests (PFTs) was the template bleeding time, in which a standardized small cut is made on the subject’s forearm and the bleeding duration timed. However, this test is invasive, labor-intensive, impractical to repeat frequently, poorly reproducible, and only modestly predictive for bleeding problems.
In vitro PFTs use various platelet agonists to activate and aggregate the patient’s platelets.7 For example, the PFA-100® device (Siemens, Munich, Germany) simulates capillary blood flow through a chamber after activation by collagen and either epinephrine or ADP. Prolonged “closure time” with collagen/epinephrine but not collagen/ADP suggests aspirin or other antiplatelet medications, whereas when both pairs are abnormal, other congenital or acquired platelet dysfunctions may be present. This type of testing is sometimes used as a screen in patients with a history suggesting platelet problems or von Willebrand disease (vWD). However, the sensitivity and specificity are low. False negatives are common, and abnormal results can also be caused by thrombocytopenia, uremia, or anemia. Several other devices test for specific antiplatelet medication effects from aspirin or P2Y12 inhibitors.7
 Platelet aggregation is the most detailed overall PFT. Platelets are tested with multiple isolated agonists to assess their patterns of physical aggregation and, in turn, the platelets’ own agonist release. Some uncommon congenital disorders lack responses to specific agonists in a characteristic fashion. More detailed testing may be needed for a specific diagnosis, such as electron microscopy for granule defects, flow cytometry for surface receptors and granule markers, or genetic testing.8
Platelet aggregation is the most detailed overall PFT. Platelets are tested with multiple isolated agonists to assess their patterns of physical aggregation and, in turn, the platelets’ own agonist release. Some uncommon congenital disorders lack responses to specific agonists in a characteristic fashion. More detailed testing may be needed for a specific diagnosis, such as electron microscopy for granule defects, flow cytometry for surface receptors and granule markers, or genetic testing.8
vWD is a factor deficiency which imparts clinical features of platelet dysfunction, due to the central role of vWF in cross-linking activated platelets to form the platelet plug.9 Up to 1% of all patients have vWD, with a wide range of severity due to either quantitative or functional defects of vWF. Diagnostic testing is integral to deciphering the specific defect and type of vWD to ensure the appropriate treatment. Since vWF is the carrier for factor VIII (FVIII) in plasma, vWF protein levels usually correlate with FVIII levels. Initial testing for vWD should include the vWF antigen level, vWF activity level, and FVIII activity level for comparison with vWF. Blood group O persons have shorter plasma half-life and lower normal levels of vWF, so ABO blood typing may be needed to interpret a borderline vWF level. Type 1 vWD is a quantitative deficiency, with decreased antigen and activity. Type 2 vWD may have normal antigen levels, but decreased activity from a defective protein. Within type 2, there are several subtypes with different molecular defects, and specialized identification is needed in order to determine the best therapy. Type 3 vWD is a rare, very severe autosomal recessive deficiency.9 The clinical features and management of vWD are discussed later in this chapter.
Laboratory Evaluation of Secondary Hemostasis and Coagulation
 A general oversight of plasma clotting factor activity is obtained by the prothrombin time (PT) for the intrinsic (tissue) pathway and the aPTT for the extrinsic (contact) pathway (Fig. 16-2), with both tests completed through the common pathway.6 These clotting tests are performed in blood specimens collected in a chelator (3.2% citrate) which binds Ca2+ to prevent clotting in the tube. The in vitro clotting test is activated by TF in the PT or negatively charged surfaces in the aPTT, using phospholipid as a platform (substituting for platelets). Ca2+ is then added to overcome the specimen chelation, and the time is measured until complete fibrin clotting is observed. Representative normal ranges are around 12 to 15 seconds for the PT and 25 to 35 seconds for the aPTT, but are defined by each laboratory using its own equipment, reagents, and normal specimens. Testing is routinely performed at 37°C, but hypothermia in the patient impairs the enzymatic reactions of clot formation.
A general oversight of plasma clotting factor activity is obtained by the prothrombin time (PT) for the intrinsic (tissue) pathway and the aPTT for the extrinsic (contact) pathway (Fig. 16-2), with both tests completed through the common pathway.6 These clotting tests are performed in blood specimens collected in a chelator (3.2% citrate) which binds Ca2+ to prevent clotting in the tube. The in vitro clotting test is activated by TF in the PT or negatively charged surfaces in the aPTT, using phospholipid as a platform (substituting for platelets). Ca2+ is then added to overcome the specimen chelation, and the time is measured until complete fibrin clotting is observed. Representative normal ranges are around 12 to 15 seconds for the PT and 25 to 35 seconds for the aPTT, but are defined by each laboratory using its own equipment, reagents, and normal specimens. Testing is routinely performed at 37°C, but hypothermia in the patient impairs the enzymatic reactions of clot formation.
Clotting physiology is more complicated than the traditional diagrams of separate cascade pathways for these two tests. We have seen that thrombin from the intrinsic pathway activates the extrinsic pathway. In vitro, the aPTT clotting test is activated by synthetic contact materials which initiate via factor XII, so deficiencies of XII and other related contact factors cause a prolonged aPTT. However, deficiencies of these contact factors do not cause bleeding and may in fact be associated with impaired fibrinolysis and thrombosis. Fibrinogen activity is also a critical parameter. Most assays measure the functional conversion of fibrinogen to fibrin, although the fibrinogen protein level can also be measured for comparison to assess fibrinogen dysfunction. Normal fibrinogen levels are around 150 to 400 mg/dL.
Mixing Studies
To investigate unexpectedly elevated PT or aPTT values, the test should be repeated after mixing the patient’s plasma with equal volumes of normal plasma. Even in severe factor deficiencies, the PT or aPTT shows substantial correction toward normal in a mixing study. However, if the patient’s plasma contains an inhibitor or an anticoagulant, the normal plasma will also be affected and the PT or aPTT will not be correct.
Individual factor level activities are determined by the degree of correction which patient plasma gives when mixed with factor-deficient plasma. The classic congenital factor deficiencies are FVIII deficiency (hemophilia A) and factor IX deficiency (hemophilia B). Both are X-linked and thus nearly always in males. Factor XI deficiency is most often seen in persons of Ashkenazi Jewish descent. Acquired factor deficiencies usually involve multiple factors.10 The vitamin K–dependent factors are II (thrombin), VII, IX, and X. In liver disease, all factor synthesis is deficient except FVIII, which also comes from endothelium. However, FVIII and other factors can be low in disseminated intravascular coagulation (DIC). As noted, FVIII may be low as part of vWD. Isolated factor X deficiency occurs in some patients with amyloidosis because the abnormal protein absorbs this factor. Performing a set of factors V, VII, and VIII may suggest a pattern to aid in the diagnosis of specific clinical syndromes.
Coagulation inhibitors are substances, usually antibodies, which block one or more clotting factors. Most do not cause bleeding; the most common examples are lupus anticoagulants (LAs), one type of antiphospholipid antibodies (APLA) discussed below in thrombosis tests. However, factor-specific inhibitor antibodies can block clotting in vivo and cause bleeding.10 They are identified by their effect on the plasma factor’s activity and semiquantified by assessing how much interference the patient’s plasma gives to factor level measurements in normal plasma. Some severe hemophiliacs and other factor-deficient patients develop alloantibodies to therapeutic clotting factors, interfering with treatment and necessitating alternative factor therapies or immunosuppression. Bovine thrombin used for topical hemostasis can induce cross-reacting antibodies to the patient’s own factor V. Autoantibodies to specific clotting factors, most commonly FVIII, can cause serious coagulopathy.
 DIC describes unchecked coagulation initiated by pathologic systemic activation of the intrinsic clotting pathway. The specific pathophysiology of DIC is discussed later in this chapter, but diagnostic criteria require an inciting condition such as extensive tissue injury or a systemic inflammatory response secondary to infection, obstetrical complication, or malignancy. Intravascular platelet activation and fibrin formation lead to thrombocytopenia, hypofibrinogenemia, and RBCs sheared by fibrin strands (schistocytes). The results of coagulation profile tests vary, but often show prolonged PT and aPTT. In some patients, thrombosis is the most prominent clinical finding, but in most patients, the depletion of platelets and clotting factors with accompanying activation of fibrinolysis leads to diffuse consumptive coagulopathy. Fibrin formation followed by fibrinolysis generates the fibrin fragments called D-dimers, which when quantified in immunologic testing are a useful indicator of DIC.
DIC describes unchecked coagulation initiated by pathologic systemic activation of the intrinsic clotting pathway. The specific pathophysiology of DIC is discussed later in this chapter, but diagnostic criteria require an inciting condition such as extensive tissue injury or a systemic inflammatory response secondary to infection, obstetrical complication, or malignancy. Intravascular platelet activation and fibrin formation lead to thrombocytopenia, hypofibrinogenemia, and RBCs sheared by fibrin strands (schistocytes). The results of coagulation profile tests vary, but often show prolonged PT and aPTT. In some patients, thrombosis is the most prominent clinical finding, but in most patients, the depletion of platelets and clotting factors with accompanying activation of fibrinolysis leads to diffuse consumptive coagulopathy. Fibrin formation followed by fibrinolysis generates the fibrin fragments called D-dimers, which when quantified in immunologic testing are a useful indicator of DIC.
Three other tests are commonly performed during surgery with whole-blood specimens: The activated clotting time (ACT), ecarin clotting time (ECT), and viscoelastic whole-blood clotting with thromboelastography. The ACT, a point of care test, assesses the intrinsic clotting pathway and is used mainly to monitor heparin anticoagulation and its protamine reversal during cardiopulmonary bypass or vascular surgery. The ECT also describes the intrinsic clotting function, but it is primarily used for measuring the clinical effects of direct thrombin inhibitors (DTIs) such as bivalirudin. These agents are often used for patients with HIT. The ACT and aPTT also reflect the clinical efficacy of DTIs, but at high doses required for cardiopulmonary bypass, ECT is more accurate.11
Thromboelastography
Whole-blood clotting and fibrinolysis can be assessed by viscoelastic testing in thromboelastography (TEG®, Haemoscope Corp., Niles, IL, USA) or rotation thromboelastometry (ROTEM®, Pentapharm GmBH, Munich, Germany). These tests measure the rate, strength, and lysis, if any, of clot formation. Numerous parameters can be measured with these tests; accordingly the TEG–ROTEM working group attempted to standardize the parameters obtained from both testing modalities in order to make them more clinically relevant. There are minor differences in the mechanisms for TEG versus ROTEM; however, both involve the use of whole blood in a heated cup with the addition of a sensor pin. The cup or the pin oscillates while the blood clots. The increasing resistance to oscillation is transmitted through the sensor pin and a graphic depiction of clot formation is displayed in the thromboelastogram.12 The patterns obtained can implicate defects in factor levels, platelet function, fibrinogen concentration, and the presence of abnormal fibrinolysis, the latter which is difficult to measure rapidly otherwise. Testing can be performed in the presence of inhibitors of heparin or fibrinolysis to help judge whether these drugs would be effective. This test format has also been adapted to assess antiplatelet therapy in patients with ventricular assist devices.13 Thromboelastography is helpful in determining the appropriate therapy, including platelets, plasma, fibrinogen replacement, or antifibrinolytics, as complex bleeding syndromes such as massive hemorrhage with consumptive or dilutional coagulopathy progress.
Diagnosis of Thromboembolic Disorders
 The risk for DVT, PE, VTE, and other thromboses is increased by intercurrent factors such as physical inactivity or immobilization, malignancy, oral contraceptives, estrogen therapy, and pregnancy. However, in addition to or especially in the absence of such factors, laboratory testing often identifies an underlying congenital or acquired predisposing abnormality which tips the hemostatic systems toward thrombosis. Discovery of one or more risk factors may influence the course of therapy and suggest benefit from family studies.
The risk for DVT, PE, VTE, and other thromboses is increased by intercurrent factors such as physical inactivity or immobilization, malignancy, oral contraceptives, estrogen therapy, and pregnancy. However, in addition to or especially in the absence of such factors, laboratory testing often identifies an underlying congenital or acquired predisposing abnormality which tips the hemostatic systems toward thrombosis. Discovery of one or more risk factors may influence the course of therapy and suggest benefit from family studies.
Congenital Risk Factors for Thrombosis
The most commonly tested congenital problems discussed below increase the risk of VTE.14 Although arterial thrombosis may involve a few of these factors, platelets are more directly involved on the arterial side, and congenital contributions are less well defined. Some investigators have described a “sticky platelet syndrome,” with hyperactive platelet aggregometry. Although severe congenital problems may present in childhood, they are rare; most thrombotic presentations are in adulthood over a lifetime of potential risk. Congenital problems are mostly categorized as deficiencies in antithrombotic pathways or hypercoagulable clotting factors.
Several congenital factors involve the protein C-ase complex and its function. The most common hypercoagulable mutation is factor V Leiden (FVL), in about 5% of Caucasians.15 FV is the cofactor for factor X when the latter activates prothrombin to thrombin. APC is the natural brake on FV, by cleaving it at Arg506. FVL carries the autosomal dominant mutation Arg506Gln, rendering FV fairly resistant to APC. Thus FV is overactive and thrombin formation is favored. The FVL polymorphism is readily identified genetically. However, a small percentage of persons with resistance to APC have other mutations in FV or other conditions. Therefore, slightly more inclusive is the functional clotting test for APC resistance, which assesses plasma clotting time with and without reagent APC.
Protein C itself is functionally deficient in up to 0.5% of the population, with autosomal dominant inheritance. This leads to overactive FVIII and FV cofactors in their respective intrinsic tenase and prothrombinase complexes. Most have low activity and antigen (type I), but some have low activity with normal antigen levels (type II). Homozygous protein C deficiency is a very severe thrombotic disorder beginning in infancy. Protein S deficiency can lead to thrombosis because of its cofactor role to protein C. Around 1 in 700 persons has autosomal dominant deficiency. Protein S circulates partly bound to the complement C4 binding protein and partly as the unbound (free) active form. Nearly all cases of protein S deficiency can be identified by assaying the free antigen and then categorized as to whether the total antigen is low (type I) or normal (type II). Type II has low function but normal antigen levels, but this is rare. Both protein C and protein S are vitamin K–dependent, and therefore vitamin K deficiency or warfarin interferes with laboratory assessment of their activities. Warfarin-induced skin necrosis in protein C or S deficiencies is discussed in antithrombotic therapy testing below. In AT-III deficiency, the relative lack of its normal blocking function on the key enzymes VIIa, IXa, Xa, and XIa leads to thrombosis risk. Testing for AT-III activity will detect both quantitative and qualitative defects.
The best characterized congenital gain in function is the prothrombin mutation G20210A (guanine to adenine). This autosomal dominant condition is found in about 1 in 50 Caucasians, but is much less prevalent in African and Asian backgrounds. Persons with this variant have high circulating prothrombin levels as the reason for thrombosis risk. Genetic testing for the mutation is more definitive than prothrombin levels. Elevated FVIII levels may be a modest risk factor for thrombosis, but FVIII is an acute-phase reactant and rises in many intercurrent conditions. Whether there is an inherited element to persistently elevated FVIII levels is unclear.
Acquired Risk Factors for Thrombosis
Several factors increase the risk of thrombosis.16 APLA are associated with both arterial and venous thrombosis risk. These antibodies bind to phospholipid–protein complexes. Several possible mechanisms for their in vivo effects have been proposed. They may bind to and activate endothelial cells, which in turn could directly tip off coagulation and/or cause vascular injury. They may interfere with phospholipids in the protein C-ase enzyme complex, leading to diminution of protein C’s regulatory function. The various antigenic targets and mechanisms of APLA require multiple tests for their detection. Studies should include tests of clotting function, most notably LA tests, and tests of solid-phase binding to antigen targets, such as anticardiolipin antibodies (ACLA) and anti-β2-glycoprotein-1 (AβGP). AβGP is a protein often present in the phospholipid–protein complex targeted by these antibodies.
LA antibodies are a common cause of prolonged aPTT which does not correct by mixing with normal plasma. It should be emphasized that the prolonged aPTT is a phenomenon of the in vitro test and is NOT associated with bleeding. However, not all LAs prolong the aPTT. Laboratories testing for LA should use at least two different tests to improve detection. One is usually a test based on the aPTT, but modified with phospholipid reagent selected to be sensitive to LA interference. A second clotting-based test is also recommended, such as the dilute Russell viper venom time (DRVVT), in which the snake venom activates factor X in the common pathway, leading to thrombin formation. This test’s reagent phospholipid is adjusted by dilution to be LA-sensitive, but because the venom bypasses the usual initiating factors, the DVVRT is not affected by autoantibody inhibitors of FVIII or other upstream factors, a potential cause of confusion in the PTT-based assays.
ACLA and AβGP antibody tests usually employ enzyme immunoassays (EIA). AβGP may be more specific for physiologic thrombotic effect, by presenting an actual target of in vivo antibodies, whereas ACLA may develop in other conditions such as infections. For example, false-positive non-treponemal syphilis serology is sometimes seen with ACLA.
Hyperhomocysteinemia is a risk factor for venous and possibly arterial thrombosis. This amino acid is made from methionine and is then either converted back or processed to cysteine. The mechanism for thrombosis risk is unclear, but endothelial cell injury has been proposed. Fasting total homocysteine is the initial screening test. Hyperhomocysteinemia can be due to various congenital mutations in homocysteine’s metabolic pathways or can be acquired via vitamin deficiencies affecting its metabolism (folate, B12, B6) or in many other medical conditions.
Monitoring Anticoagulation Therapeutic Agents
 Most anticoagulant therapies need ongoing or selective testing for assessment of therapeutic effect. Appropriate monitoring ensures that these agents are maintained within the therapeutic range; otherwise patients are at risk of thromboembolism and bleeding complications both of which can have devastating consequences.
Most anticoagulant therapies need ongoing or selective testing for assessment of therapeutic effect. Appropriate monitoring ensures that these agents are maintained within the therapeutic range; otherwise patients are at risk of thromboembolism and bleeding complications both of which can have devastating consequences.
Warfarin Anticoagulation
Warfarin therapy must be monitored by the PT and its analogue for this purpose, the international normalized ratio (INR), in order to avoid under- or overcoagulation. PT methods and reagents can widely differ between laboratories, yielding varying PT values for the same degree of factor deficiency. However, each PT test vendor supplies a conversion parameter to express the PT as the INR in patients on warfarin. The INR is a normalized value which is intended to compare results across laboratories for evaluating combined deficiencies of factors II, VII, IX, and X, the warfarin-dependent factors. The INR’s therapeutic range for warfarin anticoagulation is generally 2.0 to 3.0, except for mechanical heart valves and prevention of myocardial infarction (INR 2.5 to 3.5).17
When warfarin is started or stopped, the factors with the fastest plasma turnover, that is, the shortest half-lives, decline or rise the fastest, respectively. Thus, the inhibitor protein C, with a 6 hr half-life, declines faster than most clotting factors when warfarin takes effect, and this can cause an imbalance toward clotting during the initiation of warfarin therapy. Warfarin-induced skin necrosis is a thrombotic complication often occurring when previously unrecognized congenital protein C deficiency accentuates this imbalance.
Warfarin’s pharmacology is affected by genetic variations in the metabolism of the drug (cytochrome P450, CYP2C9) or its counterbalancing vitamin K (vitamin K epoxide reductase complex subunit 1, VKORC1). Genetic polymorphism testing has been advocated for achieving more rapid therapeutic effect when initiating therapy or in assessing difficulty achieving the target INR, but indications for genetic testing are still under investigation. The INR is not calibrated to evaluate non-warfarin deficiencies such as liver disease, which affects most other clotting factors, and thus strictly speaking, the INR should not be used in other conditions including liver disease.
Heparin Anticoagulation Testing
The aPTT is used to assess heparin anticoagulation. Each laboratory determines its own therapeutic target range for heparin anticoagulation, typically on the order of 1.5 to 2.5 times the normal mean. The laboratory determines the exact range for their test system based on a functional enzymatic test for heparin action, the anti-factor Xa activity (aFXa). (This is a different test than the factor X activity level.) Using the aFXa assay, therapeutic target heparin levels of 0.3 to 0.7 aFXa units/mL are correlated with aPTT results for that range. aFXa testing can be helpful in assessing heparin resistance.
Low-molecular-weight heparin (LMWH) drugs and their analogue, synthetic pentasaccharide (e.g., fondaparinux), do not affect the aPTT assay, and coagulation testing is usually not needed. However, if necessary, the drugs’ plasma activity levels can be assessed by the aFXa assay. This may be helpful in renal failure affecting drug excretion or in pregnant women, obese, and neonates for whom drug levels are less certain after subcutaneous injection. Like heparin, these agents inhibit factor Xa indirectly, that is, via their enhancing effect on antithrombin.
Heparin and to a lesser degree LMWH can stimulate the production of antibodies against the heparin–PF-4 complex. These antibodies can in turn cause HIT and/or activation of platelets to induce thrombosis.18 If thrombocytopenia or thrombosis develops in a patient on these drugs, tests for HIT antibodies are available by EIA or by functional measures such as serotonin release. Patients with HIT must avoid heparin and LMWH.
Several newer anticoagulants have entered clinical practice, as alternatives to heparin in patients with HIT or as alternatives to warfarin. These are direct anticoagulants which are not mediated by antithrombin. The “xaban” class of drugs, including rivaroxaban and apixaban, directly inhibit factor Xa.19 They prolong the PT and aPTT, but monitoring is not recommended and the INR should not be used. An aFXa-type assay adapted for these drugs is being researched.
DTIs also pose challenges for coagulation testing. These include hirudin from leeches, its recombinant “-rudin” mimicking molecules (bivalirudin, desirudin, lepirudin), and small synthetic molecules acting at the same site as hirudin on thrombin (argatroban, dabigatran). They all prolong the PT and aPTT and interfere with clot-based fibrinogen assays. There is no consensus on how to quantify the effect of these drugs. The ECT has been promulgated.20 Ecarin, an enzyme in snake venom, cleaves prothrombin to a metabolic intermediate which is inhibited by hirudin and its analogues. However, the ECT is not widely available.
BLOOD COMPONENT PRODUCTION
Blood Collection
The production of blood components is highly controlled by regulations and accreditation requirements in the interests of donor and recipient safety and therapeutic efficacy. Blood donors are carefully screened and tested, and blood products are made in specialized laboratories and other facilities. Regional blood centers collect and provide most blood components for transfusion, although some hospitals collect blood or platelets to augment their supply. Virtually all blood components come from volunteer unpaid donors. Pharmaceutical companies process plasma into various derivatives or synthesize some desired proteins for infusion.
Blood donors undergo a confidential interview to screen for medical problems in their donation safety and for risks of disease transmission to their recipients.21 They are questioned about risk factors, exposure, or signs of human immunodeficiency virus (HIV), hepatitis, and other infections. There are geographically based deferral criteria for tropical exposure to malaria and (in American donors) European exposure to variant Creutzfeldt–Jakob disease (vCJD). The donor’s pulse, blood pressure, and hemoglobin/hematocrit level (US minimum 12.5 g/dL or 38%) are checked. Phlebotomy is performed with validated antiseptic measures to reduce the risk of bacterial contamination in the blood bags. In the United States, donors are deferred for 8 weeks after a whole-blood donation to avoid iron deficiency.
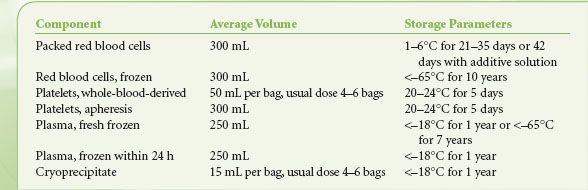
Table 16-1 shows the contents and storage parameters for blood components. In whole-blood donations, 450 to 500 mL of blood is collected into citrate anticoagulant and then separated by centrifugation into RBCs, platelets, and/or plasma. The RBC units usually have most plasma removed and replaced with preservative. In the United States, the plasma must be frozen within 6 hours of collection to be labeled fresh frozen plasma (FFP). A large proportion of plasma is now made as plasma frozen within 24 hours, with minimal effect on clotting factor content compared to FFP. Cryoprecipitate is made from barely thawed FFP, which has a precipitate enriched in fibrinogen; the precipitate is isolated by centrifugation and refrozen.22 Five bags of “cryo” comprise a typical adult dose. Whole-blood–derived platelets (sometimes called “random-donor platelets”) are derived from platelet-rich plasma in the United States and from the buffy-coat centrifugation layer in other countries.23 Four to six units are pooled to yield one adult-sized dose of platelets. Traditionally, pooling was done at the hospital just before transfusion, but blood centers can now provide pre-pooled platelets to hospitals.
TABLE 16-1. BLOOD COMPONENTS
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


