Chapter 52 Acute Respiratory Distress Syndrome in Children
The acute respiratory distress syndrome is a common and devastating clinical syndrome of acute lung injury that affects both medical and surgical patients. In 1967, Ashbaugh et al. described 12 adults with the acute onset of tachypnea and cyanosis refractory to oxygen therapy, diffuse infiltrates on the chest x-ray, and decreased lung compliance.1 Initially called the “adult” respiratory distress syndrome, this entity has now been coined the “acute” respiratory distress syndrome (ARDS), since it does also occur in children2–5 and in newborns.6 However, ARDS is rare in children, and its incidence is estimated to be from 1% to 5% of the children hospitalized in the PICU.7–10
Because the initial definition lacked specific diagnostic criteria, there was controversy over the incidence and natural history of the syndrome. Some investigators attempted to exclude cardiogenic causes of pulmonary edema by requiring a “normal” pulmonary capillary wedge pressure, and specified values range from 12 mm Hg or less,11 to less than 18 mm Hg.12 In 1988, an expanded definition was proposed that quantified the respiratory impairment through a four-point lung-injury scoring system that was based on the degree of infiltration evident on the chest radiograph, the ratio of the partial pressure of arterial oxygen to the fraction of inspired oxygen (PaO2/FiO2), the level of positive end-expiratory pressure (PEEP), and the decrease in the static lung compliance.13 Mild-to-moderate acute lung injury (ALI) was defined by a lung injury score less than 2.5, and severe lung injury (ARDS) by a score higher than 2.5. Although the lung injury scoring system has been widely used to quantify the severity of lung injury in clinical trials, it cannot be used to predict the outcome during the first 24 to 72 hours after the onset of ARDS. In contrast, when used at 4 to 7 days after the onset of the syndrome, scores greater than 2.5 may be predictive of a complicated course and the need for prolonged mechanical ventilation.
Furthermore, the direct (e.g., aspiration, fat embolism, drug ingestion, toxic gas inhalation, infectious pneumonia) or indirect (e.g., sepsis, acute pancreatitis, multiple blood transfusions, disseminated intravascular coagulation) cause of the acute lung injury was thought to influence the outcome, as well as the presence or absence of nonpulmonary organ dysfunction. However, the former concept was later challenged.14
In 1994, the American-European Consensus Conference Committee proposed a new definition (Table 52-1).15 The consensus definition had two advantages. Firstly, it recognized that the severity of clinical lung injury varies: patients with less severe hypoxemia (as defined by PaO2/FiO2 ratio ≥200 and ≤300) were considered to have ALI, and those with more severe hypoxemia (as defined by PaO2/FiO2 ratio <200) were considered to have ARDS. Earlier recognition of patients with ALI was supposed to facilitate earlier enrollment of patients in clinical trials. Secondly, the definition was simple to apply in the clinical setting, where measurement of static compliance had been largely abandoned. However, the new definition did not take into account other important factors that influence the outcome,16 such as the optimization of ventilation including peep level,17,18 a significant improvement in blood gas exchange within the first 24 hours,19 the underlying cause, and whether other organ systems were affected.20 Nevertheless, the widespread acceptance of both the 1988 lung injury scoring system and the 1994 consensus definition has improved the standardization of clinical research and trials.21 Using the 1994 definition, the radiographic diagnosis of ARDS showed a large interobserver variability in adults22 and in children.23 Also, diffuse alveolar damage with the involvement of the four lung quadrants is seen in less than half of the patients, and is not well-correlated to defined histological changes.24 Thus the evaluation of PaO2/FiO2 or SpO2/FiO2 ratios25 on the second day of optimized care has been introduced in order to better identify different degrees of lung injury of divergent outcome.26

Pathogenesis
Endothelial and Epithelial Injury
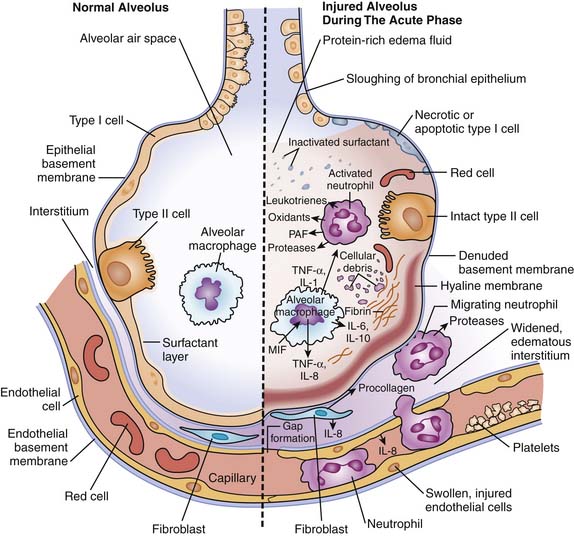
The alveolar-capillary barrier is formed of two separate cellular linear barriers, the vascular endothelium and the alveolar epithelium.27 The acute phase of ALI and ARDS is characterized by the influx of protein-rich edema fluid into the air spaces as a consequence of increased permeability of the alveolar-capillary barrier.28 The importance of endothelial injury and increased vascular permeability to the formation of pulmonary edema that characterizes the early phase of ARDS has been well established.
The critical role of epithelial injury to both the development of and recovery from ARDS has been more recently recognized.29 The loss of epithelial integrity has numerous consequences. First, the diffuse alveolar damage contributes to alveolar flooding. Second, epithelial injury disrupts normal epithelial fluid transport, and impedes the removal of edema fluid from the alveolar space. Alveolar fluid clearance has been found to be impaired in the majority of patients with ALI and ARDS.30 Third, alveolar type II cell injury impedes surfactant production and turnover, contributing to the characteristic surfactant abnormalities.31 Finally, the inflammatory cascade is activated with the release of numerous mediators. Activated macrophages secrete proinflammatory cytokines including tumor necrosis factor-α (TNF-α), and interleukins (IL-1, IL-6, IL-8) that act locally to stimulate chemotaxis and activate neutrophils.32 Neutrophils adhere to the injured capillary endothelium and marginate through the interstitium into the airspace. Neutrophils can, in turn, release oxidants, proteases, leukotrienes, and many other proinflammatory molecules (Figure 52-1).33
Role of Cytokines
A complex network of cytokines and other proinflammatory compounds is thought to initiate and amplify the inflammatory response in ALI and ARDS.34 The regulation of cytokine production may be influenced by extrapulmonary factors including microbial products, lipopolysaccharide endotoxins, and macrophage inhibitory factor, which has been found in high concentrations in the bronchoalveolar lavage fluid of patients with this syndrome.35
Not only the production of proinflammatory cytokines is important, but also the balance between the proinflammatory and antiinflammatory mediators.36 Several endogenous inhibitors of proinflammatory cytokines have been described, including soluble tumor necrosis factor receptor, interleukin-1 receptor antagonist, and antiinflammatory cytokines such as interleukin-10 and -11. The critical role of cytokines in acute lung injury and ventilator-induced lung injury has been, however, recently challenged.37
Role of Ventilator-Induced Lung Injury
While high fractions of inspired oxygen have long been considered potentially toxic to the lung, experimental evidence has recently accumulated that high volumes and pressures can injure the lung, causing increased permeability edema in the uninjured lung,38 and enhanced edema in the injured lung.39 Initial theories focused on capillary stress failure due to alveolar distension.40 More recently, alveolar distension associated with the repeated collapse and reopening of alveoli was shown to initiate a cascade of proinflammatory cytokines.41 As most patients with acute lung injury who die do so from multisystem organ failure, it has been postulated that ventilator-induced lung injury plays a key role in determining the negative clinical outcome of patients exposed to mechanical ventilation.42 The term cellular biotrauma has been coined to describe the process by which mechanical stress produced by mechanical ventilation leads to the upregulation of an inflammatory response.43 Thus cells are required to sense mechanical forces and activate opposite intracellular signaling pathways: (1) able to release growth factors and surfactant when forces are physiologic (e.g., fetal or postnatal lung breathing), and (2) proinflammatory cytokines when forces are pathologic (e.g., barotrauma or volutrauma due to either excessive inspiratory positive pressures or tidal volumes).44 Multiple other pathways can perpetuate or inhibit lung injury. Abnormalities of the coagulation system along with impaired fibrinolysis leads to alveolar fibrin formation,45 and occlusion of small pulmonary vessels by platelet-fibrin thrombi contributes to pulmonary vascular remodeling and pulmonary hypertension.46–48 Abnormalities in the production, composition, and function of the surfactant contribute also to alveolar collapse and gas exchange anomalies.49
Resolution of Lung Injury
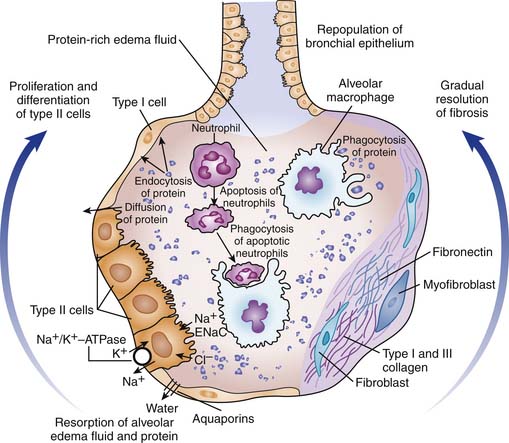
Prognosis appears dependent on resolution of the pathologic processes. Alveolar edema is resolved by the active transport of sodium and perhaps chloride from the distal air spaces into the lung interstitium (Fig. 52-2).50,51 Apical ENaC type channels and basal Na+,K+ ATPase channels appeared to be highly regulated by beta-adrenergic agents, whereas amiloride-sensitive sodium transport is modulated by basal nitric oxide.52 Water follows passively through transcellular water channels, the aquaporins, located primarily on type I cells.53 Lung fluid clearance is impaired in the majority of patients with ALI and ARDS, but maximal alveolar fluid clearance has been found to be associated with significantly lower mortality and a shorter duration of mechanical ventilation.54 The removal of insoluble protein is also particularly important, since hyaline membranes provide a framework for the growth of fibrous tissue.55 Insoluble protein appear to be removed by endocytosis and transcytosis, by alveolar epithelial cells, and by phagocytosis by macrophages.56 Type II cells proliferate to cover the denuded basement membrane and then differentiate into type I cells, restoring the normal alveolar architecture and the fluid-transport function of the alveolar epithelium. This proliferation is controlled by epithelial growth factors, including keratinocyte and hepatocyte growth factors.57 New blood vessels are formed mostly as a result of the vascular endothelium growth factor, and contribute to the normalization of the blood gas exchange.58 Potential application of mesenchymal stem cells in acute lung injury has been explored in various in vivo models.59
Fibrosing Lung Injury
After the initial phase of acute lung injury and the acute respiratory distress syndrome, progression to fibrotic lung injury usually occur by 5 to 10 days after the onset of the disorder. The alveolar space becomes filled with fibroblasts and procollagen III peptide, of which the early appearance in the alveolar space has been associated with an increased risk of death.60 The finding of marked fibrosing alveolitis on lung biopsy (or autopsy) correlates with an increased risk of death.61 The mechanisms underlying the resolution of the inflammatory-cell infiltrates and fibrosis are unclear. Apoptosis is thought to play a major role for the clearance of neutrophils from the injured lung, as high concentrations of the markers of apoptosis have been shown in the bronchoalveolar lavage fluid taken from patients with the acute respiratory distress syndrome.62,63 Antiinflammatory cytokines and proteases are also likely to play a major role. Mechanisms of repair and remodeling in acute lung injury have been recently reviewed.64 It has become apparent that the process of fibrosing alveolitis begins early in the course of ARDS.65 Determinants of persistent injury and abnormal repair and remodeling may be profoundly affected by both environmental and genetic factors. Recently, Chang et al.66 performed proteomic and computational analyses on bronchoalveolar lavage fluid in patients with ARDS to identify proteins enriched and interactions that might be important for pathogenesis of lung injury, but no clear profile has emerged yet.
Clinical Features
Incidence and Etiology
The incidence of acute lung injury and acute respiratory distress syndrome is from 1% to 5% of pediatric intensive care admissions.67–72 However, ARDS nowadays appears to be a relatively rare disease in children, perhaps because of more strict definitions or changing diagnostic criteria over the years. Within a 6-month period in nine large pediatric intensive care units across North America, 1096 (17.1%) of a total of 6403 admissions required mechanical ventilator support for a minimum of 24 hours. Of these, 701 (64%) met other criteria than primary respiratory failure for requiring mechanical support, including 13.5% upper airway obstruction, 11.5% cyanotic congenital heart disease, 9.7% life support restriction, and 5.5% chronically ventilated. In the 395 children who were eligible for respiratory failure studies, 62.4% had an acute primary diagnosis of pulmonary disease, 14.2% neurological disease, and 8.9% cardiac disease. Chronic underlying conditions were present in 43.2% of the patients. The most common acute diagnosis was bronchiolitis in infants (43.6%), and pneumonia in children 1 year old and older (24.5%). ARDS was identified in only 9 (5.7%) and 14 (9.7%) of the children with primary lung disease younger than 1 year and older than 1 year, respectively. Thus ARDS actually occurred in 23 children out of 6403 admissions, for a true incidence of 0.36%.73,74
Insults causing ALI or ARDS may either directly or indirectly injure the lung. Causes of direct lung injury include pneumonia, gastric contents aspiration, lung contusion, hydrocarbon ingestion, smoke inhalation, and sickle cell disease. Near-drowning, multiple emergent transfusions, and sepsis lead to ALI as part of multiorgan failure. Sepsis is the more common and lethal predisposing condition associated with ARDS, whether it is related to community-acquired infection (e.g., severe meningococcal shock), nosocomial infection (e.g., ventilator-associated pneumonia),75 or serious underlying conditions (e.g., immunocompromised states).76
Clinical Course
The clinical course of ALI and ARDS parallels the histopathological abnormalities. Clinical changes that first occur are tachypnea, dyspnea, agitation, and hypoxemia. These findings usually develop rapidly during a period of hours, but can evolve over 1 to 5 days.77 As the lung become edematous and consolidated, tachypnea and hypoxemia are caused by progressive restrictive lung disease and respiratory muscle fatigue. Chest radiographs show diffuse bilateral alveolar opacities, sometimes accompanied by small pleural effusion (Figure 52-3). However, CT scans reveal juxtaposition of abnormal dense lung regions and more functional lucent regions. Dense lung regions often develop in the dependent regions of the lung, reflecting collapse of the edematous lung with secondary atelectasis, whereas aerated regions usually prevail in nondependent areas.78 Lung function measurements show a reduced functional residual capacity. Total lung compliance is reduced, but compliance of the small regions of functional lung is normal. Hypoxemia results from intrapulmonary shunting and the regions with low ventilation-perfusion relationships. These regions can be recruited in response to PEEP, thereby improving oxygenation.79
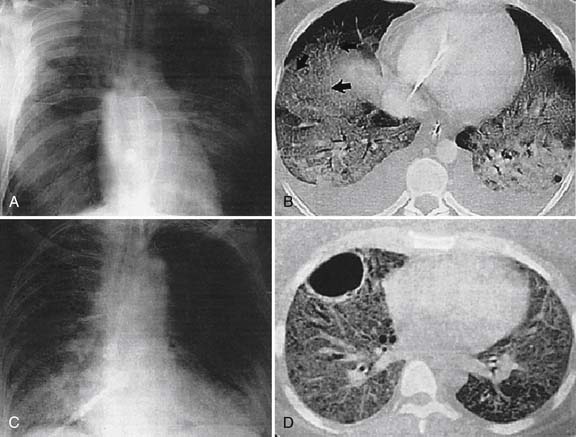
During the fibroproliferative phase, lung compliance is reduced by progressive lung fibrosis, and the effects of PEEP on oxygenation are less impressive. Lung parenchyma becomes better aerated despite accentuated interstitial markings indicative of fibrosis. At this stage, carbon dioxide retention is not uncommon. The requirement for mechanical ventilation may be prolonged for weeks, and clinical recovery usually requires a few months in children.80 Although pediatric survivors of acute hypoxemic respiratory failure perceive no limitation in lifestyle, significant abnormalities in lung function, including bronchoreactivity, have been found.81 Likewise, adult survivors of ARDS have persistent functional disability 1 year after discharge from the intensive care unit. Most patients have extrapulmonary conditions, with muscle wasting and weakness being most prominent.82
Oxygenation and Ventilator Strategy
Cellular toxicity of oxygen has been well established in both animals and human beings.83 However, high levels of oxygen appear to be surprisingly well-tolerated in patients with acute respiratory failure, even for several days.84 The reasons for this phenomenon are unclear. Antioxidant defense mechanisms have been found to be more efficient in primates than in rats.85 Furthermore, prior lung exposure to endotoxin or cytokines (i.e., tolerance phenomenon) has been shown to protect animals exposed to 100% oxygen for several days, as compared to naive animals, who died rapidly.86
Ventilator-induced lung injury has recently been the subject of intensive experimental and human research (see Chapter 51). Traditionally, mechanical ventilation was delivered in an effort to normalize arterial blood gases. Tidal volumes of 10 to 15 mL/kg of body weight were used to normalize PaCO2 values. After numerous experimental studies clearly demonstrated that excessive tidal volume traumatized lung structures, particularly when functioning lung volume is reduced to an aerated “baby lung” surrounded by extensive consolidated areas,87 an international conference addressed factors likely responsible for ventilator-induced lung injury.88 Several clinical trials evaluated lower ventilator settings as a means to minimize ventilator-induced lung injury and pulmonary fibrosis, even at the expense of normal blood-gas exchange, with little success apart from one preliminary study.89–92 The culmination of this research, the ARDS Network trial, ultimately reported convincing improved clinical outcomes with a lung-protection ventilator strategy, compared with standard treatment for ARDS, in 861 adults.93 The study group received 4 to 6 mL/kg of predicted body weight tidal volume and positive plateau pressure of less than 30 cm H2O, compared with an 11 to 12 mL/kg tidal volume and positive plateau pressure of less than 50 cm H2O in the control group. PEEP was adjusted to minimize FiO2 but maintain oxygen saturation between 88% and 95%. PaCO2 values were allowed to rise, but pH was maintained higher than 7.15. Adult patients receiving a lung protection strategy had a significantly reduced mortality rate (31% vs. 40%), fewer hospital days with extrapulmonary organ failure, and increased ventilator-free days (55% vs. 66%) over the first 28 hospital days compared with the control group). However, the marked improvement in the significant outcomes was later criticized when it appeared that the control group may have received higher than normal tidal volume on purpose.94 Indeed, the lack of difference in significant outcomes observed in all previous trials but one may have been related to the relatively limited tidal volume (~10 mL/kg) used in the control group. A fierce debate ensued and was summarized in two successive commentaries.95,96 Nevertheless, this paradigm is now also applied in children.97
PEEP maximizes alveolar recruitment, improves oxygenation insufficiency, and minimizes the need for oxygen supplementation, thereby minimizing iatrogenic oxygen toxicity. Early use of PEEP does not prevent the development or progression of ARDS.98 In animal models of ALI, tidal volumes delivered without PEEP increase the shear stress between interfacing normal and injured regions, creating neutrophilic inflammation and cytokine release similar to ARDS.99 However, PEEP can also overdistend the normal lung and compromise hemodynamic function (Figure 52-4).100 Intensivists have therefore searched for the “best” PEEP, which maximizes lung compliance and oxygenation efficiency yet minimizes overdistension and reduced cardiac output.101 For years, the issue of how to set up optimal PEEP remained controversial, when recently a new trial of the ARDS Network demonstrated that in patients with ALI and ARDS who received a tidal volume of 6 mL/kg of predicted body weight and an end-inspiratory plateau-pressure limit of 30 cm H2O, clinical outcomes were similar whether lower (8.3 ± 3.2 cm H2O) or higher (13.2 ± 3.5 cm H2O) PEEP was used throughout days 1 to 4.102 Two other trials of higher PEEP in adult patients receiving lung-protective ventilation failed to demonstrate a mortality difference but were safe and showed benefits that advocates will find reassuring.103,104 Practical guidelines for ventilator settings in ALI or ARDS are provided in Table 52-2.105
< div class='tao-gold-member'>







Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


