WEAKNESS
NICHOLAS TSAROUHAS, MD AND CHRISTOPHER F. VALENTE, MD
Weakness is defined as an inability to generate normal voluntary force in a muscle or normal voluntary torque about a joint. Although often associated, hypotonia is not always synonymous with weakness. Neurologists define hypotonia as decreased resistance to “passive” motion. Not all hypotonic patients are weak; for example, a patient with Down syndrome may have normal strength yet have decreased tone.
PATHOPHYSIOLOGY
Weakness is a reflection of a disease process that may involve any component of the motor neuron unit. These diseases are classically categorized as upper or lower motor unit disorders (Table 78.1). Upper motor neuron disease affects structures extending from the motor strip of the cerebral cortex, through the corticospinal tracts of the spinal cord, to (but not including) the anterior horn cell. Although upper motor neuron disease is generally characterized by increased deep tendon reflexes (DTRs) and spasticity, early in the clinical course there may be flaccid paralysis. Lower motor neuron disease may involve the anterior horn cell, the peripheral nerves, the neuromuscular junction (NMJ), or the muscle fibers. In general, lower motor neuron disease is associated with fasciculations, muscle atrophy, hypotonia, and hyporeflexia, and may ultimately lead to flaccid paralysis.
DIFFERENTIAL DIAGNOSIS
The cerebral cortex can be damaged by cerebrovascular accidents (CVAs), which include cerebral infarctions and cerebral hemorrhages. CVAs, while rare (2.1 to 13.1 per 100,000 children per year), cause some of the most catastrophic cases of weakness (Table 78.2). These children usually present with sudden, unilateral, or asymmetric weakness.
Cerebral hemorrhage is usually due to a ruptured arteriovenous malformation (AVM), but may also be caused by a ruptured aneurysm. Most AVMs are asymptomatic until rupture, but some children do complain of periodic “migraine-like” headaches. Brain tumor hemorrhage may also present acutely as weakness, severe headache, and vomiting.
Cerebral infarctions usually occur in the setting of predisposing factors, which include sickle cell disease, homocystinuria, structural arterial disease (e.g., moyamoya), hypercoagulable states (e.g., antithrombin III, protein C and S deficiencies, and factor V Leiden mutations). Other more common hypercoagulable states include pregnancy, malignancy, infections, and severe dehydration. Recent studies have shown increased risk associated with hypertension, diabetes, obesity, and hypercholesterolemia in younger teenage populations. Substance abuse with tobacco, alcohol, cocaine, and amphetamines have also been associated with cerebral infarction. Atherosclerosis and atrial fibrillation, common culprits in adult stroke, remain rare causes of stroke in children. Embolic causes that should be considered in pediatric patients include children with congenital heart disease, mitral valve prolapse, or a history of rheumatic fever.
Transient ischemic attacks (TIAs) often present with resolving weakness. TIAs are defined as transient neurologic deficits referable to a cerebral artery territory in a child whose cranial magnetic resonance imaging (MRI) shows no acute ischemia, but whose history/workup suggests cerebrovascular disease. Transient focal deficits are common after a seizure and are called Todd postictal paralysis. Deficits usually resolve within minutes to hours after a seizure has ended, but one study demonstrated a mean symptom duration of 15 hours and persistence of up to 36 hours. In this same study, 8/14 patients with Todd postictal paralysis had an underlying CNS lesion.
Because a Todd paralysis resolves with time, if the patient is not improving (regaining function) in 30 to 45 minutes, the differential diagnosis should be broadened to ensure a cerebrovascular event or mass lesion is not the cause of the focal weakness. Cranial imaging is crucial in cases of prolonged postictal paralysis, especially if the mental status remains impaired, or if other focal deficits persist. The reference standard study is MRI. However, access to MRI may be limited or may not be immediately practical or safe (e.g., need for deep sedation). Consequently, a computed tomography (CT) scan of the head is often the initial study of choice. The head CT is most useful to rule out acute bleeds and large mass lesions. In some cases, a CT angiogram is necessary to identify vascular lesions or anomalies.
Traumatic injuries may seriously damage or compress the spinal cord. Spinal cord concussion is defined as a transitory disturbance in spinal cord function caused by a direct blow to the back. Symptoms may include flaccid paraplegia or quadriplegia, a sensory level at the site of injury, loss of tendon reflexes, and urinary retention. Recovery usually begins within a few hours and is usually complete within a week. Spinal epidural hematoma may cause spinal cord compression as the hematoma expands. Emergent spinal MRI scanning is indicated when an epidural hematoma is suspected. Other traumatic injuries include vertebral body compression fractures, dislocations, and spinal cord transections.
Another serious cause of spinal cord compression is epidural abscess, which is usually caused by hematogenous spread of bacteria, most commonly Staphylococcus aureus, or by direct spread from an adjacent carbuncle or vertebral osteomyelitis. This is a rare (0.2 to 1.2 per 10,000 hospital admissions), but potentially devastating disease entity often unidentified until progression to severe neurologic sequelae. Patients commonly present with fever and back pain, but may also have headache, vomiting, stiff neck, and bowel and bladder dysfunction. Point tenderness may be elicited over the affected area. The diagnosis is confirmed by MRI, which helps also to distinguish the abscess from a vertebral discitis. Similarly, spinal cord tumors are another important cause of spinal cord compression.
TABLE 78.1
DIFFERENTIAL DIAGNOSIS OF WEAKNESS
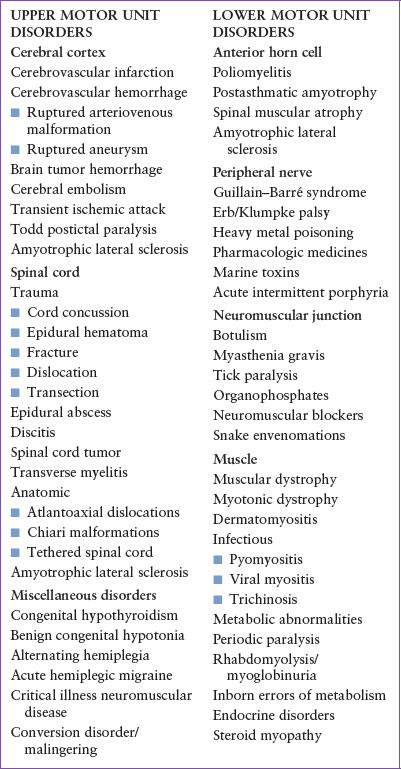
TABLE 78.2
LIFE-THREATENING CAUSES OF WEAKNESS

Transverse myelitis is an acute demyelinating disorder of the spinal cord. It is frequently attributed to a preceding viral infection, but may be immune mediated. It presents as an acute episode of fever and back pain at the level of cord involvement. The hallmark of transverse myelitis is the demarcation of the lesion by a sensory loss below a spinal cord level (usually thoracic). Leg paresthesias and weakness evolve rapidly over the course of 2 days. Asymmetric leg weakness is common. Tendon reflexes may be increased or reduced. Bowel and bladder continence are often lost. Urgent MRI of the spine is required to exclude cord compression.
Anatomic anomalies of the spine and spinal cord associated with weakness also include the atlantoaxial dislocations associated with Klippel–Feil and Down syndrome. Patients with Chiari malformations/myelomeningoceles also have weakness (as well as other deficits). In the growing child, a tethered spinal cord may cause weakness and neurologic deficits, as the tether causes the spinal cord to stretch. Clumsiness may be the presenting symptom of leg weakness. Bladder control problems are also common.
Juvenile amyotrophic lateral sclerosis (ALS) is a rare hereditary disorder involving upper and lower motor neurons. Similar to “adult” ALS, or Lou Gehrig disease, it causes spasticity and muscular atrophy. The course is progressive and is ultimately fatal.
Anterior horn cell disease affects the most proximal component of the lower motor neuron unit. Because these diseases affect the motor neurons, sensory function is normal. Reflexes are generally lost early in the course of the disease. Ultimately, muscle atrophy and fasciculations develop. Cranial nerve nuclei are often affected as well.
Poliomyelitis is the classic example of an anterior horn cell disease, but it has been largely eradicated by immunization. In 2014, anterior horn cell disease emerged in association with enterovirus D68. A peculiar entity that mimics poliomyelitis is idiopathic postasthmatic amyotrophy, or Hopkins syndrome. It presents as a sudden onset of weakness, generally 1 to 2 weeks after an acute asthma attack. Like polio, prognosis is poor, with all patients left with some degree of permanent paralysis.
The three pediatric types of spinal muscular atrophy (SMA) comprise a group of autosomal recessive genetic disorders in which the anterior horn cells in the spinal cord and motor nuclei of the brainstem are progressively lost. There is widespread muscle denervation and atrophy. The weakness is usually symmetric, progressive, and proximal, and presents anytime from birth to adulthood. Later in the disease course, tongue fasciculations and diminished DTRs may occur. Importantly, intellect and cardiac, sensory, and sphincter functions are preserved.
Spinal muscular atrophy I (acute infantile SMA, or Werdnig–Hoffman disease) is the most severe form of SMA. The weakness and severe generalized hypotonia begin before 6 months of age. These patients can never sit alone. Death often occurs by 4 years of age, usually from overwhelming pneumonia. Spinal muscular atrophy II (chronic infantile SMA) usually has its onset of weakness between 6 and 18 months. These patients can sit alone, but can never walk. In this “intermediate” SMA, survival to adulthood is expected. Spinal muscular atrophy III (mild juvenile SMA, or Kugelberg–Welander disease) usually presents with weakness after 18 months. These patients can walk without support.
Neuropathies (primary disorders of the axon or its myelin sheath) usually present as progressive symmetric distal weakness. Weakness and sensory loss may move in a “glove and stocking” fashion. Tendon reflexes are usually lost early. Dysesthesias (“pins and needles” or burning sensations) usually occur in acquired conditions.
Guillain–Barré syndrome (GBS), or acute inflammatory demyelinating polyradiculopathy (AIDP), is the classic acquired immunologic neuropathic disorder. It is the most common cause of acute motor paralysis in children. GBS occurs when activated immune mechanisms, induced by an antecedent viral infection, trigger inflammation and demyelination (see Chapter 105 Neurologic Emergencies). Many viruses have been implicated, including adenovirus, Epstein–Barr virus, cytomegalovirus, human immunodeficiency virus, varicella-zoster virus, measles virus, Rubulavirus (mumps), and vaccinia virus. Mycoplasma pneumoniae and Campylobacter jejuni infections, as well as some vaccines (i.e., swine flu 1976), have also been implicated.
The most common complaint is weakness, but patients also present with leg and back pain, and in younger children, an abnormal gait. The symptoms may progress for days to weeks. The weakness is usually symmetric and may be ascending or descending. There is often a sensory loss, as well as loss of position and vibratory sense. DTRs are diminished or absent in the weak muscles. Bowel and bladder incontinence, autonomic dysfunction (hypotension), and cardiac dysrhythmias also occur.
Respiratory paralysis occurs in 20% to 30%. Cranial nerve involvement is seen in 30% to 40% of patients, usually manifested by facial weakness or ocular paresis. The Miller Fisher variant of GBS includes the triad of ataxia, areflexia, and ophthalmoplegia.
Required clinical criteria for diagnosis include a progressive motor weakness of more than one limb, and areflexia. Examination of the cerebrospinal fluid demonstrates an elevated protein without pleocytosis (albuminocytologic dissociation). Important diseases to consider in the differential diagnosis include acute cerebellar ataxia, transverse myelitis, toxic neuropathy, tick paralysis, botulism, myasthenia gravis (MG), and acute viral myositis. A chronic form of GBS, chronic inflammatory demyelinating polyneuropathy (CIDP), also exists. The course of CIDP varies widely and ranges from complete spontaneous recovery to frequent relapses with gradually declining function. Therapies for both GBS and CIDP include steroids, IVIG, plasmapheresis, and physiotherapy.
Birth trauma may produce traction injuries to the nerve roots causing a restricted pattern of focal weakness. Erb palsy, a proximal (C5/C6) brachial plexus palsy, is the most common brachial plexus injury. These infants assume the “waiter’s tip” posture: arm adducted, humerus internally rotated, elbow extended, forearm pronated, wrist flexed. Klumpke palsy, an injury to the lower trunk (C8/T1) of the plexus, is much rarer. This typically manifests as an infant holding the arm in supination with the elbow flexed and wrist extended. Fortunately, most birth-related traction injuries of the brachial plexus result in only a self-limited neuropraxia from stretching of the nerves, with resolution of symptoms by 6 months of age. More extensive brachial plexus injuries can be associated with Horner syndrome (ptosis, miosis, anhidrosis) due to injury of the stellate ganglion. Avulsions or ruptures of the nerves usually result in more permanent weakness. Flaccid weakness of the arms and legs may result from excessive traction on the spinal cord during a difficult delivery.
Heavy metals, such as lead, mercury, arsenic, and thallium, are known neuropathic toxins. Lead, in particular, may cause a distal motor weakness with foot and wrist drop. Drug-induced neuropathies occur with drugs of abuse, as well as pharmacologic medicines. Several antimicrobials (isoniazid, nitrofurantoin, and zidovudine), as well as antineoplastics (vincristine, vinblastine, cytosine arabinoside, and cisplatin), are known to cause paresthesias and muscle weakness.
The seas also harbor deadly neurotoxins. Ciguatera and paralytic shellfish poisoning are caused when toxin-elaborating dinoflagellates are ingested by fish or shellfish, which are then eaten by unsuspecting humans. Cone snail stings, blue-ringed octopus bites, sea snake bites, and puffer fish ingestion may also result in life-threatening paralysis. On land, the Crotalidae (pit vipers) and Elapidae
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


