5 Very High Systemic Arterial Blood Pressure
The Joint National Committee (JNC) on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure has defined two acute conditions of elevated systemic arterial pressure.1 A hypertensive emergency is characterized by the presence of elevated systemic blood pressure (BP) and new or progressive end-organ damage, including but not limited to the cardiac, renal, and central nervous systems. A hypertensive emergency is an infrequent clinical situation that requires immediate BP reduction (not necessarily to normal ranges). Although the absolute BP elevation is not a criterion for the diagnosis, a hypertensive emergency is typically associated with a diastolic blood pressure (DBP) above 120 mm Hg. If unrecognized or left untreated, hypertensive emergencies can lead to acute myocardial infarction (MI), pulmonary edema from left ventricular (LV) dysfunction, hypertensive encephalopathy (HE), intracranial hemorrhage, microangiopathic hemolysis, and/or acute renal failure (Box 5-1).
Box 5-1
Hypertensive Emergencies
LV, Left ventricular.
Using JNC definitions, hypertensive crises (urgency and emergency) account for more than 25% of all patient visits to a medical section of an emergency department (ED), with hypertensive emergencies accounting for one-third of the cases.2 Central nervous system (CNS) complications are the most prevalent organ system dysfunction, followed by cardiovascular dysfunction. The incidence of the disorder has remained stable at 2 to 3 cases per 100,000 population over many decades, although the prognosis associated with aggressive medical management has improved significantly.3 Most commonly, hypertensive emergencies occur in the setting of uncontrolled or unknown chronic hypertension. Hypertensive emergencies also may develop as secondary hypertension in association with such diverse etiologies as renal vascular disease, sleep apnea, hyperaldosteronism, pheochromocytoma, and pregnancy (preeclampsia).4 Postoperative hypertension occurs most often following vascular surgery procedures in patients with a background history of hypertension. Untreated postoperative hypertension can contribute to postoperative bleeding in addition to the recognized complications of hypertensive emergencies.
Additional terms used by clinicians to describe very high systemic arterial BP include accelerated hypertension, which is a severely elevated BP associated with retinal findings of ocular hemorrhages and exudates. The term malignant hypertension includes severe hypertension with the presence of ocular hemorrhages and exudates with papilledema (grade IV Kimmelstiel-Wilson retinopathy). Vascular injury to the kidney in this setting is termed malignant nephrosclerosis. The term hypertensive emergency is preferred, as end-organ dysfunction can occur in the patient with hypertension in the absence of retinal findings.5,6
 Pathophysiology
Pathophysiology
An acute elevation in systemic arterial BP most fundamentally involves an increase in systemic vascular resistance. This increase in vascular resistance is attributed to a complex interaction of circulating and local vascular mediators. Vasoconstriction is promoted by circulating catecholamines, angiotensin II (ATII), vasopressin, thromboxane (TxA2), and/or endothelin 1 (ET1). In contrast, compensatory production of local counterregulatory vasodilators, including nitric oxide (NO) and prostacyclin (PGI2), is inadequate to maintain homeostatic balance. This unregulated vasoconstriction promotes further endothelial dysfunction. A proinflammatory response, incorporating cytokine secretion, monocyte activation, and up-regulated expression of endothelial adhesion molecules, appears to occur in hypertensive emergencies, leading to promotion of endothelial hyperpermeability and activation of coagulation cascades.7 This cascade of intravascular events leads to the characteristic pathologic findings of obliterative vascular lesions. These vascular changes, evident to the clinician by examination of the retina, are mirrored by changes in the kidney, leading to a proliferative arteritis, and in advanced stages of the process, fibrinoid necrosis. Relative ischemia results in affected organs, leading to end-organ dysfunction. Early control of elevated BP is critical to prevent progression to a more advanced stage of the disease process.
Aggressive control of elevated systemic arterial BP must be undertaken with caution, however. The potential adverse effects of aggressive BP control have been most carefully considered in the cerebral circulation. Normally, cerebrovascular arteriolar tone is adjusted over a range of cerebral perfusion pressures in order to maintain a constant cerebral blood flow (CBF). Increases in cerebral perfusion pressure (CPP) promote an increase in vascular resistance, whereas decreases in CPP act to vasodilate the cerebral vasculature. In normal individuals, constant flow is therefore maintained over a range of mean arterial pressure (MAP) from approximately 60 mm Hg to 150 mm Hg.8 As MAP increases to values over 180 mm Hg, or the upper limit of autoregulation, cerebral hyperperfusion can occur, resulting in cerebral edema. Conversely, when CPP falls below the lower limit of autoregulation, CBF decreases, and tissue ischemia may occur. In patients with long-standing hypertension, a rightward shift of the CPP-CBF relationship occurs such that the lower limit of autoregulation occurs at a value higher than in normal subjects.9 Comparative studies in hypertensive and normotensive patients suggest that the lower limit of autoregulation is about 20% below the resting MAP for both, although the absolute value is higher for hypertensive patients.10 These data support the common recommendation for a maximum BP reduction in the acute setting of 20% to 25% of the MAP from the highest values, or a DBP goal typically in the 100 to 110 mm Hg range. This regulated level of BP reduction should maintain critical organ perfusion even for patients with long-standing hypertension.
 Cerebrovascular Disease
Cerebrovascular Disease
Hypertensive Encephalopathy
Acute elevations in systemic arterial BP can lead to HE, resulting from a failure of the upper level of cerebral vascular autoregulation. The most common clinical manifestations include headache, nausea and vomiting, visual disturbances, focal neurologic findings, or seizures. If left untreated, the condition can progress to coma and death. The majority of patients with HE will have a MAP significantly above the patient’s baseline BP, although not always in the range typically associated with hypertensive emergency. Retinal findings including arteriolar spasm, exudates, hemorrhages, and papilledema are often present but are not required to establish the diagnosis. Magnetic resonance imaging (MRI) studies show a characteristic edema pattern involving the subcortical white matter of the parietooccipital regions; this finding is termed posterior leukoencephalopathy.11 Best appreciated on T2-weighted images, posterior structures including the cerebellum, brainstem, and occasionally the cortex also can be affected. The findings typically are bilateral but can be asymmetric. The electroencephalogram (EEG) can show loss of the posterior dominant alpha rhythm, generalized slowing, and posterior epileptiform discharges, which resolve after appropriate therapy.12
Acute Stroke
Hypertension is present in as many as 80% of patients with acute stroke, particularly in patients with preexisting hypertension. The incidence is higher among patients with primary intracerebral hemorrhage as compared to ischemic disorders.13 The acute high systemic arterial BP most frequently declines to normal within 48 hours of presentation. The relationship between BP and mortality in patients with stroke may be “U-shaped.” According to this notion, systolic BP (SBP) values above or below 140 to 180 mm Hg are associated with increased mortality. In the International Stroke Trial, SBP above 200 mm Hg was associated with an increased risk of recurrent ischemic stroke (50% greater risk of recurrence), while low BP (particularly <120 mm Hg) was associated with an excess number of deaths from coronary heart disease.14
A number of important clinical features complicate the management of hypertension in acute stroke. First, during acute stroke, cerebral autoregulation may be compromised in ischemic tissue, and lowering of BP may further compromise CBF and extend ischemic injury. Second, medications used to treat hypertension can lead to cerebral vasodilation, augmenting CBF and leading to progression of cerebral edema.15 Ideally a “correct” level of MAP should be maintained in each patient to maintain CPP without risking worsening cerebral edema or progression of the lesion, but the clinical determination of this “ideal” value is often difficult.
A Cochrane review of 12 trials comparing an active intervention to placebo/control with 1153 total participants concluded that insufficient evidence existed to favor altering BP in acute stroke.16 Using available information, most consensus guidelines recommend that BP not be treated acutely in patients with ischemic stroke unless the hypertension is extreme (SBP >220 mm Hg or DBP >120 mm Hg) or the patient has active end-organ dysfunction in other organ systems.17 When treatment is indicated, cautious lowering of BP by approximately 15% during the first 24 hours after stroke onset is suggested. Antihypertensive medications are restarted approximately 24 hours after stroke onset in patients with preexisting hypertension who are neurologically stable, unless a specific contraindication to restarting treatment exists. Requiring special consideration are patients with extracranial or intracranial arterial stenosis and candidates for thrombolytic therapy. The former group is dependent on perfusion pressure so BP therapy may be further delayed. In contrast, before lytic therapy is started, treatment is recommended so that SBP is 185 mm Hg or less and DBP is 110 mm Hg or less. Blood pressure should be stabilized and maintained below 180/105 mm Hg for at least 24 hours after intravenous lytic therapy.17
Two recent clinical trials have suggested aggressive BP reduction limits hematoma expansion without clear benefit on mortality.18,19
 Cardiovascular Disease
Cardiovascular Disease
Acute Left Ventricular Dysfunction
The vast majority of patients presenting with acute heart failure are hypertensive on initial assessment.20 Hypertension can be the inciting event, with secondary myocardial dysfunction; or alternatively, hypertension can be a secondary component of acute pulmonary edema due to the sympathoadrenal response to hypoxemia, increased work of breathing, and anxiety. Efforts to control elevated systemic arterial pressure in this setting are essential because high systemic arterial BP in the patient with acute pulmonary edema contributes to increased myocardial workload and diastolic dysfunction. In contrast, the use of vasodilators in patients with acute pulmonary edema and normal to low BP can have deleterious effects.21,22 Similar to the patient with cerebrovascular disease, a U-shaped blood pressure/mortality relationship is expected.
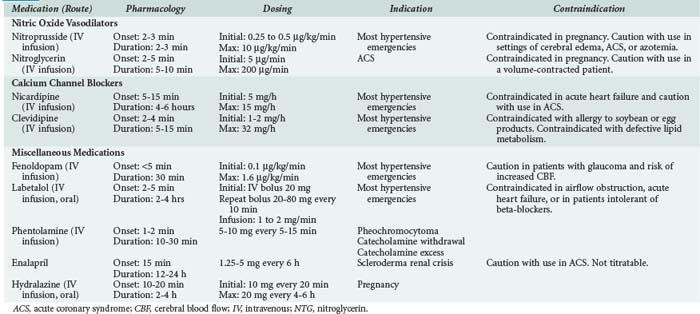
In addition to the more traditional IV vasodilators, IV calcium channel antagonists have demonstrated efficacy in the treatment of acute hypertension in the setting of LV dysfunction. The dihydropyridine calcium channel antagonists nicardipine and clevidipine can reduce systemic arterial pressure while preserving coronary blood flow.23 Fenoldopam, a dopamine-1 receptor antagonist, also has been has been shown to preserve coronary blood flow during treatment to reduce systemic arterial pressure in this setting.24 Despite their demonstrated efficacy in the treatment of hypertensive emergency, limited data exist with these newer agents to suggest superiority over NTG or nitroprusside. Agent selection should first be influenced by the adverse risk profile associated with the individual agents (Table 5-1). When not contraindicated by specific risk, the agents with a more favorable cost profile (i.e., NTG, nitroprusside) should be used based upon equivalent efficacy.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


