Ventilator-Induced Diaphragmatic Dysfunction: Introduction
Controlled mechanical ventilation (CMV) is a ventilator mode where the respiratory muscles are not contracting, and the ventilator takes full responsibility for inflating the respiratory system. Evidence accumulating over the past two decades has shown that CMV can induce dysfunction of the diaphragm, mainly consisting of atrophy, oxidative stress, and ultrastructural injury, resulting in decreased diaphragmatic contractility. This is called ventilator-induced diaphragmatic dysfunction (VIDD).1
The frequency with which CMV is used cannot be determined with certainty. An international survey revealed that 13% of mechanically ventilated patients receive a neuromuscular blocker for 8% of the total days of ventilator support.2 In these patients, full ventilator support is mandatory. Moreover, it was recently shown that the use of neuromuscular blockers during the first 2 days of ventilator support improves the survival of patients with severe acute respiratory distress syndrome,3 a finding that may lead to increased use of CMV. Other patient groups not receiving neuromuscular blockers also receive full ventilator support, such as patients with traumatic brain injury, postoperative neurosurgical patients, comatose patients, patients with status epilepticus on barbiturate coma to suppress seizure activity, and so on. Thus, a considerable percentage of ventilated patients are at risk of developing VIDD.
Evidence for Ventilator-Induced Diaphragm Dysfunction in Humans
The first human evidence for the existence of VIDD came from retrospective analysis of postmortem data obtained in neonates who received ventilatory assistance for 12 days or more immediately before death. This study revealed diffuse diaphragmatic myofiber atrophy (small myofibers with rounded outlines), not present in extradiaphragmatic muscles or diaphragms of infants ventilated for 7 days or less.4
The Components of Ventilator-Induced Diaphragm Dysfunction
Ventilator-induced atrophy7 is usually measured by the reduction of the cross-sectional area of myocytes in histologic sections. Atrophy has been observed in both slow-twitch and fast-twitch human diaphragmatic fibers and is quite significant (Fig. 43-1), its magnitude amounting to 40% to 50% after quite variable periods of CMV (range: 15 to 276 hours.)5,6,8 Atrophy preferentially affects the diaphragm because it was not observed in the pectoralis major muscle of the same patients.5 The longer the duration of CMV, the greater the observed atrophy (Fig. 43-2).6
Figure 43-1
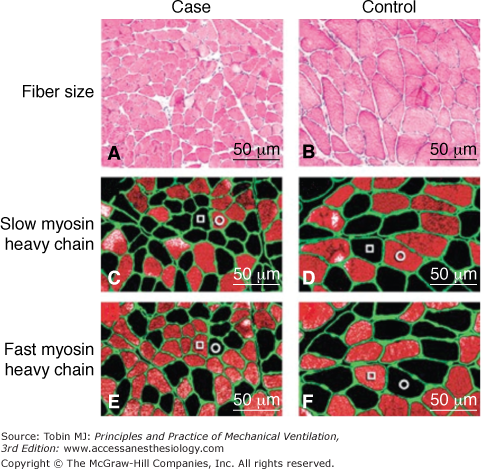
Atrophy in the human diaphragm after CMV. The slow-twitch and fast-twitch fibers in the case specimens (Panels A, C, and E) are smaller than those in the control diaphragms (Panels B, D, and F). Panels A and B (hematoxylin and eosin) show that neither inflammatory infiltrate nor necrosis is present in case or control specimens. The sections in Panels C and D were preincubated with NOQ7.5.4D antibody, which is specific for the slow myosin heavy chain, whereas sections in Panels E and F were preincubated with the MY-32 antibody, which reacts with all fast myosin heavy chains. In addition, in each section, all fibers are outlined by an antibody reactive to laminin. In each of the sections, fibers reacting with the antibody appear orange-red, whereas fibers not reacting with the antibody appear black. In Panels C, D, E, and F, a representative slow-twitch fiber is indicated by an open circle and a fast-twitch fiber by an open square. (Used, with permission, from Levine et al.5)
Figure 43-2
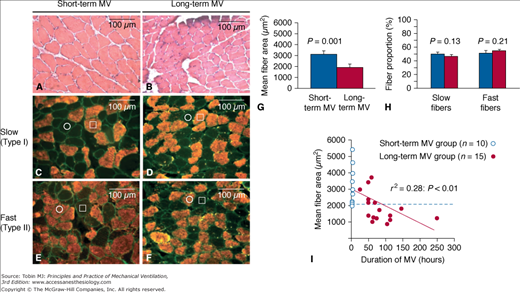
Relationship between duration of mechanical ventilation (MV) and diaphragmatic atrophy. Representative images of transverse frozen sections obtained from the diaphragms of the short-term MV (A,C,E) and long-term MV (B,D,F) groups are shown. The diaphragmatic sections are stained with hematoxylin and eosin (A, B) or with antibodies directed against slow (C, D) or fast (E, F) isoforms of myosin heavy chain. C to F. Serial sections, individual representative slow-twitch and fast-twitch fibers are marked by an open square and circle, respectively. G and H. Quantitative analyses of diaphragmatic fiber size (mean cross-sectional area) and fiber-type proportions in the two patient groups, respectively. I. The significant correlation between the degree of diaphragmatic atrophy and the duration of MV (horizontal dashed line indicates lowest value of fiber size measured in short-term MV group). (Used, with permission, from Jaber et al.6)
Animal studies also suggest that atrophy is more pronounced in the diaphragm, which atrophies earlier than the peripheral skeletal muscles, which are also inactive during CMV.9–11 Two days of CMV with positive end-expiratory pressure (PEEP) (2 cm H2O) induced atrophy in rabbits,12 whereas 3 days of CMV without PEEP were inadequate to induce atrophy13 in the same species, which suggests that the rapidity of atrophy development might be augmented with use of PEEP. The increased lung volume at the end of expiration (with use of PEEP) places the passive diaphragm in a relatively shortened position and skeletal muscle atrophies faster when shortened.14,15
Atrophy can result from decreased protein synthesis, increased proteolysis, or both. Six hours of CMV in rats decreased the in vivo rate of mixed muscle protein synthesis (averaged for all muscle proteins) by 30% and the rate of myosin heavy-chain protein synthesis by 65%, both of which persisted throughout 18 hours of CMV.16 In addition, 24 hours of CMV suppressed the messenger RNA levels of insulin-like growth factor-1, which stimulates protein synthesis.17 Thus, CMV decreases protein synthesis in the diaphragm.
Increased proteolysis has been documented in diaphragmatic strips of animals subjected to 18 hours of CMV.9 Mammalian cells have four different protein systems and organelles for proteolysis—the calpains,6 the caspases,5 the proteasome system,5,8 and the autophagy-lysosomal system8—all of which are activated in the human diaphragm after CMV, a finding previously suggested by animal models.9,18–20
Calpains, which are activated after CMV (Fig. 43-3), do not fully degrade, but only partially cleave proteins in vivo. This renders the proteins amenable to the proteasome.7 The stimulus for calpain activation in not known, but calcium elevation in the cell is a prerequisite. The reduced (messenger RNA) levels of sarcoplasmatic reticulum calcium adenosine triphosphatase (ATPase; the enzyme that removes calcium from the sarcoplasm), secondary to 24 hours of CMV21 in animal models may contribute to calpain activation.
Figure 43-3
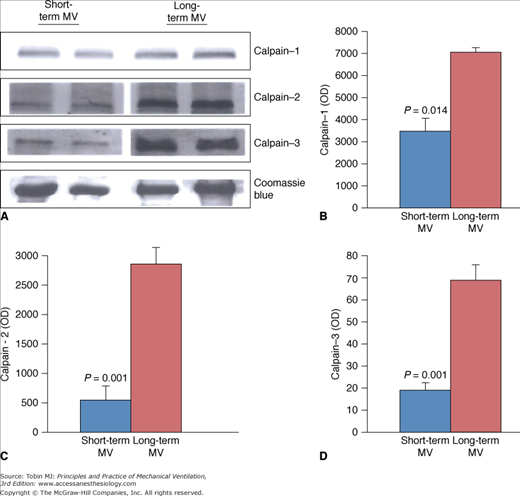
Expression of calpain isoforms in diaphragms of short-term and long-term mechanical ventilation (MV) groups. Representative immunoblots (A) and group mean quantification of protein levels measured in diaphragmatic tissues obtained from the short-term and long-term MV groups for calpain-1 (B), calpain-2 (C), and calpain-3 (D). (Used, with permission, from Jaber et al.6)
Caspases are proteases that can degrade proteins and especially complexes of actin and myosin18,22 and release them from the myofibrillar lattice. Upregulation of caspase-3 expression has been documented in the diaphragm secondary to CMV in both humans5 and in animal models of VIDD.19 Caspase-3 can be activated by oxidative stress, increased intracellular calcium, and increased calpain activity.18
In animal models of VIDD, the decreased volume of the cytoplasm (atrophy) was observed in the presence of decreased number of myonuclei (skeletal muscle cells are multinucleated cells and theoretically a single myonucleus can sustain the necessary gene expression for a limited area of the cytoplasm, a relationship known as the myonuclear domain23), so that the myonuclear domain remains constant.19 This decrease in myonuclear content was mediated by a caspase-3 dependent increase in apoptosis, which was evident as early as 6 hours after the onset of CMV.19 Both the apoptosis and atrophy were attenuated with caspase-3 inhibition.19
CMV-induced apoptosis has also been documented in the human diaphragm (Fig. 43-4).24 The finding of elevated caspase 9, but not caspase 8, and elevated Bcl2-interacting mediator of cell death (Bim) (and its transcriptional variants), but not Fas or Fas Ligand, suggest that the intrinsic (mitochondrial), rather than the extrinsic apoptotic pathway, is the primary pathway that operates in CMV-induced apoptosis in the human diaphragm, resulting in caspase 3 activation and nuclear DNA fragmentation.24 Complementary experiments, conducted in vitro, suggest that oxidative stress is a potential proximal activator of all of the pathways that have been implicated in VIDD, with mitochondrial dysfunction representing a central component of this cascade.24 Bcl2-interacting mediator of cell death is probably an important mediator of the oxidative stress-induced activation of the intrinsic apoptotic pathway, and Fos, FoxO1, and Stat3 are the transcription factors that regulate its expression.24
Figure 43-4
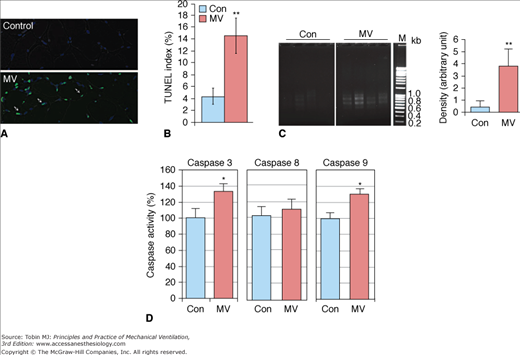
Apoptosis in the human diaphragm after CMV. CMV results in DNA fragmentation and activates caspases 3 and 9 in human diaphragm. A. TUNEL (TdT [terminal deoxynucleotidyl-transferase]-mediated [2′-deoxyuridine 5′-triphosphate]-biotin nick-end labeling) staining was performed on cryosections of control and ventilated human diaphragm. Fragmented genomic DNA was labeled with FITC (fluorescein isothiocyanate) conjugated dUTP (deoxyuridine triphosphate); positive signals appear green. Myonuclei are visualized by DAPI (4,6-diamidino-2-phenylindole) staining (blue). Note that the positive TUNEL staining signals (green) are localized in nuclei stained by DAPI (blue). B. TUNEL-positive nuclei were counted and normalized to total myonuclei; ratio is shown as the TUNEL index. Control (Con), n = 9; MV, n = 10. C. DNA fragmentation was measured by polymerase chain reaction (PCR)-based detection and visualized by electrophoresis in 2% agarose gel. Density of the PCR products was quantitated with Image J and normalized to the total DNA input. M, DNA markers. Control, n = 5; MV, n = 8. D. Caspase 3, 8 and 9 enzymatic activities from control and MV human diaphragm lysates were measured by fluorometric assay. Results are presented as relative fluorescence units after normalization to total protein amount. Control, n = 7; MV, n = 9. *, P < 0.05; **, P < 0.01. (Used, with permission, from Tang et al.24)
The autophagy–lysosome pathway is also upregulated in the ventilated human diaphragm.8 Autophagy (Greek for self-eating) is a catabolic pathway characterized by the formation of vesicles (autophagosomes) that engulf cytoplasmic organelles and proteins, which then fuse with lysosomes that degrade their contents. This process is a major mechanism for degrading long-lived proteins and organelles. Autophagy occurs at low basal levels to perform homeostatic functions, but can be rapidly upregulated when cells need to generate energy. Hussain et al8 demonstrated that prolonged CMV triggers the appearance of autophagosomes (Fig. 43-5) and increases the expression of autophagy-related genes in the diaphragm (Fig. 43-6). This is associated with upregulation of the transcription factor FOXO-1. It should be emphasized that these changes are relatively specific to the diaphragm and are not observed to the same degree in the control limb muscle (quadriceps) of the same patients. Moreover, upregulation of the autophagy-related genes in the diaphragm increases with the time spent on CMV.8
Figure 43-5
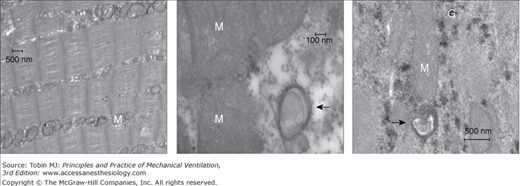
Left panel. Representative electron micrograph of a section of a diaphragm from a control subject showing normal ultrastructure and absence of autophagosomes. Middle and right panels. Representative electron micrographs of a section of a diaphragm from a brain-dead organ donor undergoing controlled mechanical ventilation. These sections show autophagosomes (black arrows) in close proximity to mitochondria (M). G indicates glycogen particles. (Used, with permission, from Hussain et al.8)
Figure 43-6
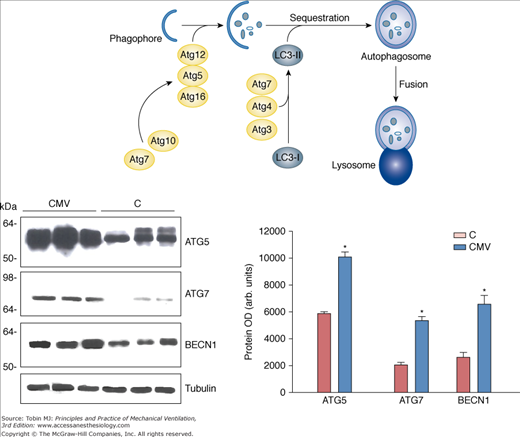
Autophagy-related genes in the human diaphragm after controlled mechanical ventilation (CMV). Top. Schematic cartoon of autophagosome assembly. Left. Representative immunoblots of BECN1, ATG5, and ATG7 in diaphragms of the control (C) and CMV groups. Right. Mean values of protein optical densities (OD) of BECN1, ATG5, and ATG7 in diaphragms of the C and CMV groups. *, P < 0.05 compared with control subjects; Atg, Autophagy related genes; LC3-I, LC3-II, microtubule-associated protein light chain-3 (LC3)-I or LC3-II; BECN1, beclin-1. (Used, with permission, from Hussain et al.8)
The proteasome is a multisubunit, multicatalytic complex that exists in two major forms (Fig. 43-7): The core 20S proteasome can be free or bound to a pair of 19S regulators to form the 26S proteasome, which degrades (in an adenosine triphosphate [ATP]-dependent manner) proteins covalently bound to a polyubiquitin protein chain (ubiquitinated proteins). The binding of ubiquitin to protein substrates requires the ubiquitin-activating enzyme (E1), which utilizes ATP-derived energy to form a covalent link with a ubiquitin protein, followed by transfer of the active ubiquitin moiety to a ubiquitin-conjugating enzyme (E2), and finally transfer of this ubiquitin to the protein to be degraded via a ubiquitin ligase (E3) (Fig. 43-8). CMV increases the level of ubiquitin-protein conjugates in the diaphragm,6,25 which are the substrates of the 26S proteasome (Fig. 43-9). Key enzymes involved in the function of ubiquitin-proteasome pathway are upregulated in the human diaphragm after CMV, such as the skeletal muscle specific ubiquitin ligases (E3 enzymes), muscle atrophy F-box (MAFbx/Atrogin-1), and muscle ring finger-1 (MuRF1),5,8,25 as well as the E2 conjugases UBC2 and UBC48 (Fig. 43-10). The changes in the E3 ligases are relatively specific to the diaphragm and were not observed in the quadriceps (control limb muscle) of the same patients, which were also inactive (Fig. 43-10).8
Figure 43-7
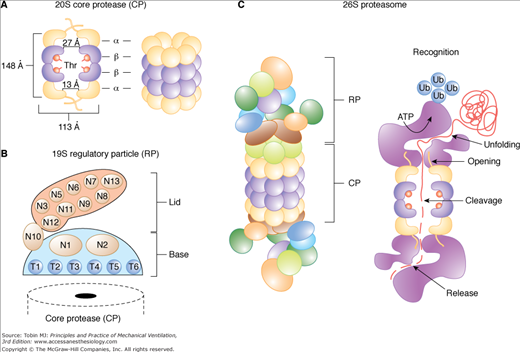
The 20S and 26S proteasome isoforms. A. Organization of the core protease (CP) and (B) the regulatory particle (RP) with its Lid and Base subparticles. The dimensions of the CP were obtained from the crystal structure of the yeast complex. The N-terminal threonine residues that form the protease active sites in the β1, β2, and β5 subunits are indicated. C. Proposed structure and sequence of events that lead to the degradation of a ubiquitinated protein by the 26S holoprotease. ATPase, adenosine triphosphatase; N, RP non-ATPase subunits; T, RP AAA-ATPase subunits; Thr, threonine; Ub, ubiquitin. (Used, with permission, from Vierstra et al.72)
Figure 43-8
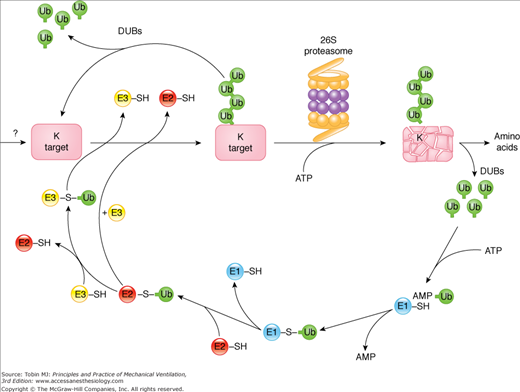
The ubiquitin–proteasome pathway of proteolysis. Proteins degraded by the ubiquitin–proteasome pathway are first conjugated to ubiquitin (Ub). The process of linking ubiquitin to lysine residues in proteins destined for degradation involves the activation of ubiquitin by the E1 enzyme in an adenosine triphosphate (ATP)-dependent reaction. Activated ubiquitin is transferred to an E2 carrier protein and then to the substrate protein, a reaction catalyzed by an E3 enzyme (E3 ligase). This process is repeated as multiple ubiquitin molecules are added to form a ubiquitin chain. Once a conjugate is assembled bearing a chain of multiple Ubs, it is either recognized by the 26S proteasome and degraded in an ATP-dependent process or the conjugate is disassembled by deubiquitinating enzymes (DUBs), releasing the ubiquitin and target intact. In ATP-dependent reactions, ubiquitin-conjugated proteins are recognized and bound by the 19S complex, which releases the ubiquitin chain and catalyzes the entry of the protein into the 20S core proteasome. Degradation occurs in the core proteasome, which contains multiple proteolytic sites within its two central rings. The ubiquitin is not degraded but is released and reused. ADP, adenosine diphosphate; K, lysine; SH, sulfhydryl. (Used, with permission, from Vierstra et al.72)
Figure 43-9
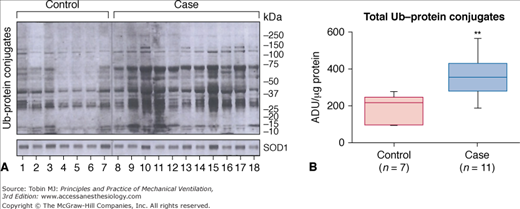
Ubiquitin–protein conjugates in human diaphragmatic cytoplasm fractions after controlled mechanical ventilation (CMV). A. Immunoblotting results for ubiquitin (Ub)–protein conjugates in seven control and eleven case specimens. The fact that the control diaphragms show ubiquitination of many bands is consistent with the concept that ubiquitination is a normal process for protein turnover. The superoxide dismutase (SOD) 1 protein-loading control did not differ between lanes. B. Case diaphragms exhibited approximately twice the amount of ubiquitinated proteins, suggesting that case diaphragms are exhibiting an upregulation of ubiquitination. **, P < 0.01; ADU, arbitrary density units. (Used, with permission, from Levine et al.25)
Figure 43-10
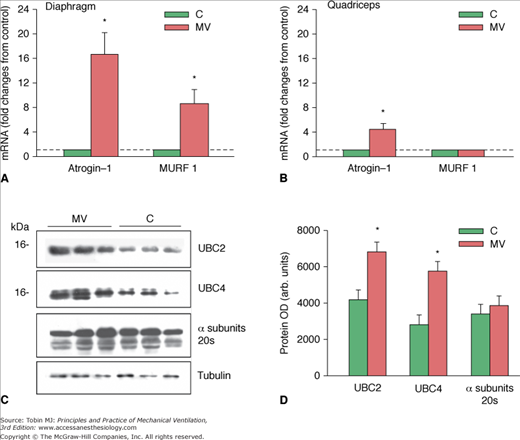
Proteasome ligase expression after controlled mechanical ventilation (CMV). A. Changes in messenger RNA (mRNA) expression levels of Atrogin-1 and MURF1 in the diaphragms of control (C) subjects and subjects undergoing CMV. *,P < 0.05 compared with control subjects. B. Changes in mRNA expression levels of Atrogin-1 and MURF1 in the quadriceps muscles of control subjects and subjects undergoing CMV. *,P < 0.05 compared with C subjects. C. Representative immunoblots of UBC2, UBC4, and a subunits of the 20S proteasome in diaphragms of the C and the CMV groups. D. Mean values of protein optical densities (OD) of UBC2, UBC4, and a subunits of the 20S proteasome in diaphragms of the C and CMV groups. *, P < 0.05 compared with control subjects. (Used, with permission, from Hussain et al.8)
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


