Simon M. Helfgott, Derrick J. Todd, Kevin Wei The vasculitides include a diverse group of uncommon disorders characterized by immune system–mediated inflammation and the destruction of blood vessel walls. Clinical signs and symptoms result from the subsequent impairment of blood flow through these damaged blood vessels to the distal tissues and organs. Patients with a vasculitic syndrome often have multiorgan dysfunction. Accordingly, these conditions are serious and often fatal if they are not recognized early and treated aggressively, usually with long-term immunosuppressive therapy. Although the specific vasculitides typically affect only certain organs, virtually any organ may be involved. Because the vasculitic syndromes are uncommon and data on disease incidence are imprecise, conclusions about the epidemiology of these conditions are difficult to make. For most conditions, age at onset is variable, ranging from infancy to old age. Some vasculitides, however, affect only certain age groups; for example, giant cell arteritis (GCA, or temporal arteritis) does not occur in individuals younger than 50 years (see Chapter 216), Takayasu arteritis is virtually unknown in patients older than 40 years, and Kawasaki disease is a vasculitis of childhood. Ethnic differences among vasculitides are less distinct; GCA tends to affect individuals of northern European ancestry, whereas there appears to be a higher incidence of Takayasu arteritis in women of East Asian descent. Ideally, classification of the vasculitides would be based on the underlying disease mechanisms of the various disorders. Our understanding of the immunopathogenesis of the vasculitides, however, remains far from complete. For this reason, vasculitides are often classified according to the size of the involved blood vessels (Box 220-1).1 This classification scheme does not necessarily account for disease mechanisms, but it is practical in that the clinical features of vasculitic syndromes that involve similar-sized vessels tend to affect common organ systems. Large-vessel vasculitides include GCA, the most common of all systemic vasculitides, and Takayasu arteritis. These diseases typically involve the aorta or its major branches, including the carotid, vertebral, subclavian, mesenteric, renal, and iliac arteries. Clinically, these syndromes manifest as large-territory claudication or infarction of the involved vascular tree. The medium-vessel vasculitides include the antineutrophil cytoplasmic antibody (ANCA)–associated vasculitides Wegener granulomatosis (WG), Churg-Strauss syndrome (CSS), and microscopic polyangiitis (MPA) as well as polyarteritis nodosa (PAN) and Kawasaki disease. The last two conditions can cause aneurysms of the involved arteries, which may supply the heart, kidney, gastrointestinal tract, and gonads. The many types of small- and medium-vessel vasculitides cause ischemia at the level of the small arteries, arterioles, or capillaries and typically affect the skin, kidneys, lung, gastrointestinal tract, and nervous system as well as other sites that tend to be more disease specific. The pathophysiology of the vasculitides is varied. Multinucleated giant cells may be observed in biopsy specimens of the temporal artery in patients with GCA and are the hallmark of this diagnosis. Hepatitis B virus (HBV) surface antigen has been found in the serum of some patients with PAN, suggesting a role for this virus in the pathogenesis of PAN.2 Infection with hepatitis C virus (HCV) can lead to the formation of cryoglobulins, which are immune complexes that are deposited in blood vessel walls and cause vascular inflammation and destruction.2 The small- to medium-vessel vasculitides, WG, CSS, and MPA, are associated with the ANCA autoantibody. The role of ANCA, if any, in the pathogenesis of these conditions has not been fully elucidated. Necrotizing granulomas are a histologic feature of WG and CSS. Unique to CSS are the large numbers of circulating and tissue-based eosinophils. Patients with a drug-induced vasculitis may have immune complex formation consisting of the foreign protein (offending drug) and its binding antibody.3 In some instances, this may represent a true serum sickness reaction in which the patient’s immune system generates antibodies against foreign protein (historically antisera). Alternatively, some medications (e.g., propylthiouracil [PTU], hydralazine, minocycline, and allopurinol) cause a vasculitis that is associated with positive ANCA serology, often in a very high titer. The contribution of ANCA in the pathogenesis of drug-induced vasculitis is unclear. Patients with connective tissue diseases such as rheumatoid arthritis, systemic lupus erythematosus, or Sjögren syndrome may develop a small-vessel vasculitis, although it is not clear how the underlying disease process in these cases contributes to the systemic vasculitis. Clinical manifestations of systemic vasculitis are protean, and therefore diagnosis and treatment are often delayed. In general, most vasculitic syndromes have characteristic clinical, laboratory, or pathologic features that allow their identification and classification.1 A systematic approach to the vasculitides, based chiefly on clinical presentation and physical examination findings, can expedite the process. Constitutional symptoms, including fever, malaise, fatigue, anorexia, weight loss, arthralgias, and myalgias, are almost uniformly present and therefore do not distinguish among the vasculitides. Their absence should call into question the diagnosis of vasculitis altogether. When fever is present, the temperature rarely exceeds 38.9° C (102° F), and it is not typically associated with rigors or chills. GCA is the prototypical large-vessel vasculitis. It is the most common of all vasculitides, although it is found almost exclusively in patients older than 50 years (see Chapter 216).4 The presentation of GCA is varied and includes the new onset of headache, scalp tenderness, cranial pain, visual disturbances, or jaw claudication in an older adult patient with constitutional symptoms. Arm claudication from subclavian artery stenosis can occur. Patients might report arm pain when brushing their hair, and a subclavian bruit may be present. Blindness is a dreaded complication of unrecognized GCA and, if it is not treated immediately, is often irreversible. Fever of unknown origin is a less common presentation. Aortic aneurysms occur, usually in the ascending thoracic aorta. Involvement of large vessels below the diaphragm (e.g., celiac, mesenteric, renal, and iliac arteries) is rare. Approximately half of patients with known GCA also report diffuse shoulder and pelvic girdle achiness and carry the diagnosis of polymyalgia rheumatica.5 On the other hand, GCA occurs in only about 10% to 20% of patients with known polymyalgia rheumatica.6 Takayasu arteritis is another large-vessel vasculitis that primarily affects women younger than 50 years.7 Patients with this vasculitis classically do not have palpable pulses, and there may be signs of unequal blood pressures, arm claudication, or bruits in the neck or arms. Other presentations include cerebral vascular accident, heart failure, and ruptured aortic aneurysm. Because these are extremely unusual events in young persons, Takayasu arteritis should be considered in the appropriate clinical setting. PAN is defined as a necrotizing arteritis that predominantly affects medium but also small arteries. Destruction of the blood vessel wall causes the development of aneurysms, which interfere with blood flow to affected organs. In addition to constitutional symptoms, common features of PAN include ulcerating skin lesions, hypertension from renal involvement, postprandial abdominal pain from mesenteric artery involvement, testicular or ovarian pain from gonadal artery involvement, and peripheral neuropathy (see later). Some patients with PAN have chronic infection with HBV.8 Kawasaki disease is a medium-vessel vasculitis of infancy and childhood classically causing high fever, bilateral conjunctivitis, tender cervical lymphadenopathy, desquamating rash, peripheral edema, and strawberry tongue. Affected children are at risk for coronary aneurysms, the major cause of death. Aspirin and intravenous immune globulin constitute the definitive therapy. Primary angiitis of the central nervous system (PACNS) is a very rare vasculitic condition that exclusively affects the brain or spinal cord, usually of older adults.9 Patients typically are seen with subacute dementia and personality changes. PACNS is often not recognized until late into the disease course, when stroke and coma occur. Brain biopsy is often required to confirm the diagnosis. Understandably, this condition is associated with significant morbidity and mortality. WG, also known as granulomatosis with polyangiitis, is a small- to medium-vessel ANCA-associated vasculitis. Although it is varied in presentation, the classic triad of involvement is that of the upper airway, lower airway, and kidneys.10 Disease of the ears, nose, and throat is common and can manifest as recurrent sinusitis, mucosal ulcerations, hearing loss, and epistaxis. Hemoptysis from pulmonary hemorrhage and stridor from subglottic stenosis often herald lower airway involvement, and both can be life-threatening. Renal involvement usually requires laboratory analysis to be identified (see later). Additional manifestations of WG can include palpable purpura, mononeuritis multiplex (defined later), central nervous system disease, and, less commonly, ocular, gastrointestinal, skeletal muscle, joint, or cardiac involvement. Overt joint swelling (i.e., arthritis) is uncommon. More often, patients complain of nonspecific arthralgias and myalgias. CSS, also known as eosinophilic granulomatosis with polyangiitis (EGPA) is an ANCA-associated small- to medium-vessel vasculitis characterized by asthma and eosinophilia.11 The condition should be suspected when a middle-aged or older adult patient develops new-onset asthma, which often precedes the vasculitis by up to 3 years. CSS vasculitis frequently affects the upper and lower respiratory tract, the peripheral nervous system as mononeuritis multiplex, and the heart as myocarditis or coronary arteritis. Other common involvement includes the central nervous system, gastrointestinal tract, skeletal muscle, joints, skin (palpable purpura), and kidney (glomerulonephritis). MPA is a third ANCA-associated small- to medium-vessel vasculitis that has only recently been established as an entity distinct from PAN. Unlike WG and CSS, which cause granulomatous vasculitis, MPA is characterized by a necrotizing inflammation of involved vessels. Features of MPA include palpable purpura, glomerulonephritis, necrotizing alveolitis, mononeuritis multiplex, and gastrointestinal disease. Certain medications and drugs may induce ANCA-associated vasculitis, including PTU, hydralazine, minocycline, and levamisole, a common adulterant found in cocaine. Symptoms from drug-induced ANCA-associated vasculitis include constitutional symptoms, arthralgias, and cutaneous vasculitis. Severe organ manifestations, including glomerulonephritis and alveolar hemorrhage, can also occur. Drug-induced ANCA-associated vasculitis is characterized by high-titer autoantibodies, including antibody to elastase or to lactoferrin. Providers who see patients taking these medications or toxins should be aware of the potential for the development of ANCA-associated vasculitis.3 Cutaneous vasculitis or leukocytoclastic angiitis typically involves the smallest blood vessels in the skin. In most cases, the skin is the only organ involved, although more widespread involvement may be observed in more severe cases. Patients are seen with palpable purpura of the lower extremities, but purpura may develop over the trunk and chest as well (see Chapter 63). Henoch-Schönlein purpura (HSP) is a small-vessel vasculitis that may affect people of all ages, but usually from early childhood to middle age. Typically, there is involvement of the joints (with pain or swelling), skin (palpable purpura), gastrointestinal tract (abdominal pain, bleeding), and kidneys (microscopic hematuria, rising serum creatinine concentration). Although this is usually a benign disorder in children, it may be a more serious condition in adults, in whom renal involvement is more likely to lead to end-stage renal disease.12 Cryoglobulinemic vasculitis occurs in the setting of patients with serum cryoglobulins. Patients often have chronic infection with HCV, although cryoglobulins can occur in other settings (i.e., malignant disease, chronic infection, and systemic rheumatologic disease). Cryoglobulinemic vasculitis classically causes glomerulonephritis, arthralgias, palpable purpura, and peripheral neuropathy. Gastrointestinal and pulmonary involvement also occurs, although rarely. In the evaluation of most small-vessel vasculitides, the presence of palpable purpura (see Chapter 63) is perhaps the most helpful physical examination finding (see Fig. 63-1). These lesions are usually multiple, more often seen in distal lower extremities. They occur as a result of extravasation of red blood cells and white blood cells outside of the destroyed blood vessel wall of the small arterioles. They vary in size from a few millimeters to 1 to 2 cm ( The small- and medium-vessel vasculitides can involve a peripheral nerve, and a careful neurologic examination may detect these findings (e.g., sensory dysesthesia, motor weakness, optic neuritis). Classic findings include wristdrop, footdrop, or a facial droop. Vasculitic involvement of a single peripheral nerve is termed mononeuritis. Mononeuritis multiplex describes the cumulative involvement of multiple peripheral nerves over time and is highly suggestive of a vasculitis in the proper clinical setting.
Vasculitis
Definition and Epidemiology
Classification of Vasculitic Syndromes
Pathophysiology
Clinical Presentation and Physical Examination
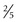 to
to 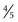 inch) in diameter. On occasion, they are pruritic. Other skin lesions, including livedo reticularis, nonpalpable purpura, and cutaneous ulcers, can also be seen.
inch) in diameter. On occasion, they are pruritic. Other skin lesions, including livedo reticularis, nonpalpable purpura, and cutaneous ulcers, can also be seen.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


