Chapter 45
Thrombocytopenia
Platelets are cell fragments derived from megakaryocytes in the bone marrow. Platelets initiate hemostasis by adhering to damaged vessels, forming a plug, and providing a surface for the coagulation cascade. Platelet counts normally vary between 150,000/μL and 450,000/μL. Platelet counts significantly < 150,000/μL constitute thrombocytopenia. The differential diagnosis of thrombocytopenia includes disorders resulting from (1) increased platelet destruction (on an immune basis or nonimmune basis), (2) decreased platelet production, and (3) sequestration of circulating platelets (Box 45.1).
Over 50% of patients in the intensive care unit (ICU) have decreased platelet counts, and most patients experience a platelet nadir around day 4 of the ICU stay. Increased mortality is seen in patients who develop moderate or severe thrombocytopenia (platelet counts < 50,000/μL), especially in patients whose thrombocytopenia persists for a prolonged time period.
Disorders of Increased Platelet Destruction by Nonimmune Mechanisms
Thrombotic Thrombocytopenic Purpura
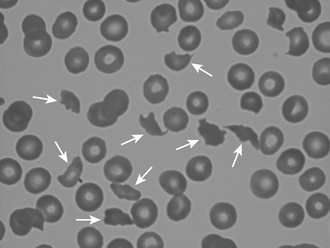
The prototypic disease for nonimmune platelet destruction is thrombotic thrombocytopenic purpura (TTP) (see Chapter 63). TTP is caused either by a congenital deficiency of von Willebrand factor cleaving protease or by auto-antibodies directed against the protease. The protease is encoded by the gene designated as ADAMTS 13. Patients with TTP present with the clinical pentad of fever, renal abnormalities, central nervous system disorders, thrombocytopenia, and microangiopathic hemolytic anemia (MAHA). MAHA is characterized by the presence of fragmented red cells, or schistocytes (Figure 45.1), caused by erythrocyte destruction in the microvasculature. Hemolytic uremic syndrome (HUS) shares pathologic features with TTP, but in HUS renal abnormalities are more pronounced. HUS is most often secondary to infection with Shiga toxin, producing organisms such as Escherichia coli O157:H7. The primary treatment for TTP is plasmapheresis (also known as apheresis) accompanied by glucocorticoids. The treatment of HUS is usually supportive. Patients with TTP become thrombocytopenic from activation of platelets caused by the presence of large von Willebrand factor multimers. This leads to intravascular platelet aggregation and ultimate clearance of circulating platelets from the circulation. Platelet transfusions in TTP have been associated with fatal outcomes, presumably from microthrombosis, and are therefore contraindicated except for life-threatening bleeding. Fortunately, patients with TTP rarely bleed. Thrombocytopenia resulting from MAHA in conditions other than TTP usually responds to treatment of the underlying condition or removal of the offending drug (Box 45.2; see also Chapter 63). Again, as with classic TTP, there should be a reluctance to transfuse platelets in all but the most serious bleeding episodes.
Disseminated Intravascular Coagulation
DIC results from various causes, including gram-negative and gram-positive bacterial infections, trauma, snakebites, brain injury, and burns. In DIC, the generation of thrombin occurs without its subsequent neutralization, which is normally carried out by coagulation pathway inhibitors. Because thrombin promotes the conversion of fibrinogen to fibrin, microvascular thrombosis occurs. This results in tissue ischemia along with consumption of coagulation components such as platelets, fibrinogen, and prothrombin. DIC is often identified clinically by skin hemorrhage in the form of petechiae and ecchymoses. Alternatively, DIC can present as thrombosis of digits. Shock, organ dysfunction, and frank hemorrhage may occur as well. Other laboratory findings in DIC include prolongation of the prothrombin time, partial thromboplastin time, and thrombin time, a decrease in the fibrinogen level, and an increased level of fibrin degradation products (FDPs), also known as fibrin split products (FSPs). d-Dimers result from the degradation of cross-linked fibrin by plasmin, and their levels also increase in DIC. Primary fibrinogenolysis, a rare condition that results when plasmin is generated in the absence of DIC, is characterized by normal levels of d-dimers despite a low fibrinogen level.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


