INTRODUCTION AND PATHOPHYSIOLOGY
Sickle cell disease is a spectrum of blood disorders of which sickle cell anemia (SCA) is the most serious. SCA is found primarily in children of African, Arab, or Mediterranean descent. It is caused by a single amino acid substitution in the sixth position of the β-globulin chain of normal adult hemoglobin (HbA), which creates the abnormal sickle hemoglobin (HbS). The HbS chain found in patients with SCA creates a hydrophobic region of the hemoglobin tetramer when it is deoxygenated.1 In this state, noncovalent polymerization of the hemoglobin molecules creates chains that distort the shape of the membrane, causing the characteristic sickle appearance. The altered red blood cell shape and its associated rigidity and decreased deformability cause sickle cells to impede blood flow. This is responsible for most of the clinical manifestations of SCA through two primary mechanisms: hemolytic anemia and vaso-occlusion with subsequent ischemia-reperfusion injury. Interruption of blood flow created by the abnormal cells leads to poor tissue perfusion, acidosis, and hypoxia, which cause further sickling. The need to reverse these conditions is central to the management of SCA-related complications.
Children homozygous for this genetic abnormality have two genetic copies of the HbS mutation and are described as having classic hemoglobin SS (HbSS) disease, whereas heterozygotes for HbS with a second β-globulin chain that is normal (HbA) are considered to have sickle cell trait. These patients have enough HbA to prevent polymerization of the hemoglobin chains and subsequently avoid the more severe complications of the disease. Other patients may be heterozygous for HbS coupled with a second hemoglobin abnormality such as HbC, HbO, or β-thalassemia. Although some of these combinations can cause the severe pathology of SCA, patients with HbSC and the 90% of HbS/β-thalassemia patients with some normal β chains have fewer ischemic and infectious complications than patients with HbSS.
Because fetal hemoglobin (HbF) is unaffected by these genetic mutations, symptoms of SCA do not appear until HbF is replaced by the abnormal HbA. HbF predominates until 4 months of age, after which it rapidly declines, reaching baseline low levels just before 1 year of age. One of the first organs affected by the emerging sickle cells is the spleen. Recurrent splenic infarcts lead to a gradual decline in splenic function between 4 and 12 months of age, resulting in susceptibility to serious infections with encapsulated bacteria.
In the United States, all neonates are screened for SCA using highly sensitive and specific tests, so the diagnosis is rarely made in the ED. The clinical manifestations and potential complications of SCA, however, include symptoms that overlap considerably with diseases not related to SCA. Therefore, the diagnostic portion of this chapter is organized in a symptom-based fashion, and the treatment portion is grouped by disease physiology.
SICKLE CELL ANEMIA SYMPTOMS
Vaso-occlusive crises causing severe extremity pain are the most common manifestation of SCA, accounting for 79% to 91% of ED visits.2 Common triggers include stress, cold, dehydration, altitude, hypoxia, or illness. Pain preferentially affects the long bones and lower back. Individual patients tend to manifest their pain crises in characteristic locations, but the frequency of vaso-occlusive pain crisis is variable, even among SS homozygotes. Five percent of children with SCA account for 33% of total episodes, averaging between 3 and 10 pain crises per year.3 Children display variable degrees of tenderness over affected sites and may have slight temperature elevations without true fever. Because anemia can precipitate a crisis, assess patients for an acute drop in hemoglobin; experienced families may know the patient’s baseline values.
Severe vaso-occlusive crises can lead to bony infarction. Pain episodes caused by bony infarcts typically cause more debilitating and refractory pain than past episodes, and there is often significant localized bone tenderness and an elevated peripheral WBC count. Fat embolism can be a complication. Signs and symptoms of fat embolism include acute respiratory distress, hypoxia, and systemic symptoms of petechiae, altered mental status, liver damage, and renal failure. Fat embolism can occur without acute extremity pain, and the aforementioned symptoms should prompt consideration of fat embolization.
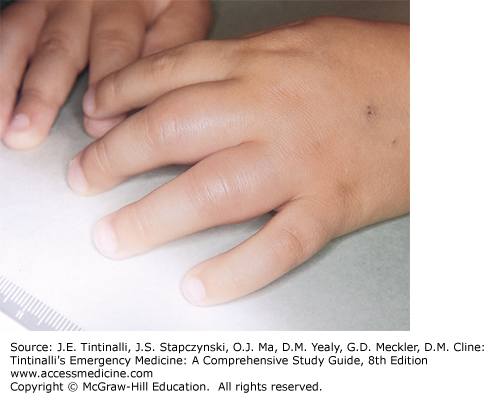
When vaso-occlusive crises affect the nutrient arteries of the metacarpals and/or metatarsals, dactylitis results, and children develop tender, painful, swollen hands and feet, which may be accompanied by a low-grade fever (Figure 142-1). Dactylitis occurs in children <2 years old and may be the initial clinical presentation of SCA, but dactylitis is extremely rare beyond 5 years of age. Any number of extremities can be affected, and symptoms can last from days to 2 weeks. Recurrences are common.
FIGURE 142-1.
Acute sickle dactylitis. Bilateral cylindrical swelling of soft tissue of the fingers in sickle cell anemia consistent with vaso-occlusive crisis of the hands. [Photo contributed by Donald L. Rucknagel, MD, PhD. Reproduced with permission from Knoop K, Stack L, Storrow A, Thurman RJ: Atlas of Emergency Medicine, 3rd ed. © 2009, McGraw-Hill, Inc., New York.]
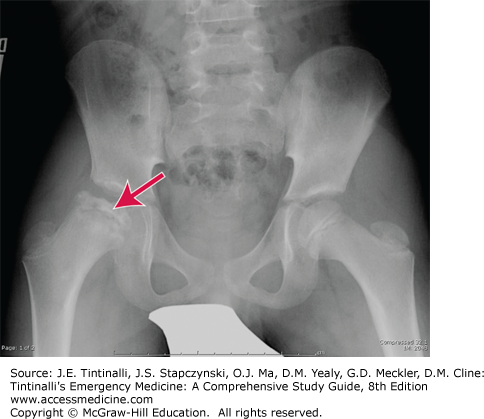
Avascular necrosis of the femoral head occurs in 30% of patients by age 30 years old. Children present with afebrile inguinal pain with weight bearing. Radiographs may demonstrate flattening and collapse of the femoral head (Figure 142-2). Less commonly, ischemia can cause similar pain in the humeral head.
Bone and joint infections affect patients with SCA at higher rates than the general population. Infections are often difficult to distinguish from bony infarcts. Symptoms of tenderness, warmth, and swelling may be indistinguishable from those of a vaso-occlusive crisis, although a high fever is more typical of infection. Limited range of motion of a joint is unusual in vaso-occlusive crisis and should prompt evaluation for a septic joint. Although leukocytosis occurs with vaso-occlusive crisis, a left shift is unique to infection. Erythrocyte sedimentation rates are unreliable in patients with SCA. Radionuclide imaging or MRI may distinguish infection from infarct.
Abdominal pain is a difficult symptom to assess in any child and is complicated by the potential abdominal manifestations of SCA. Any acute abdominal condition, such as appendicitis, pancreatitis, and hepatitis, must be considered. However, the abdomen is the next most common site of pain from a vaso-occlusive crisis after the extremities and back. Abdominal vaso-occlusive crises are due to mesenteric, splenic, and hepatic ischemia and present as sudden onset of poorly localized abdominal pain. Physical examination may reveal tenderness and guarding but typically should not demonstrate rebound or rigidity.
Gallstones are common due to the elevated bilirubin levels from hemolysis in SCA and can occur as early as 2 to 4 years of age. Diagnosis is by US. Although symptoms in children may resolve with time, occasionally, cholecystitis can be severe, and infection can progress rapidly (see chapters 79, “Pancreatitis and Cholecystitis,” and 131, “Gastrointestinal Bleeding in Infants and Children”). Right upper quadrant syndrome due to intrahepatic cholestasis is not common. Signs and symptoms include sudden right upper quadrant pain, jaundice, anorexia, tender hepatomegaly, and, often, fever. Bilirubin levels can be as high as 50 milligrams/dL. Although patients with SCA often have bilirubin levels above baseline, bilirubin levels rarely exceed 4 milligrams/dL and are predominantly unconjugated bilirubin. Confirm the diagnosis by US. Acute hepatic sequestration is another cause of abdominal pain, and the liver enlarges rapidly. Laboratories are variable, except for low hemoglobin. US or CT scan shows diffuse hepatomegaly in hepatic sequestration.
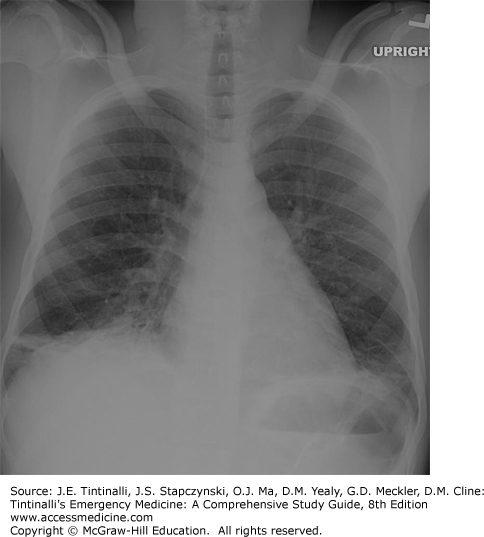
Acute chest crisis, due to pulmonary ischemia and infarction, is frequently a complication of pneumonia (Figure 142-3). The incidence of acute chest crisis in children is higher than in adults (21 per 100 person-years in patients with HbSS), but is associated with better outcomes. Children develop any combination of pleuritic chest pain, cough, fever, and dyspnea. Examination findings include retractions, hypoxia, tachypnea, rhonchi, or rales, although dehydration can minimize the presence of rales on examination. An elevated WBC count is often present in both infarct and pneumonia, and thrombocytopenia may accompany severe crises. Chest radiograph is indicated in any patient with chest symptoms or hypoxia, although findings may be minimal early on and should not dissuade treatment of a patient with potential chest crisis on clinical grounds only. Radiography demonstrates a new alveolar pulmonary infiltrate, which is typically located in the upper or middle lobe in children, although adults often have multilobar disease. The cause of pneumonia is typically Chlamydia, Mycoplasma, viral, Streptococcus pneumoniae, Staphylococcus aureus, or Haemophilus influenzae. Sputum or blood cultures are rarely positive and not generally recommended, except in patients ill enough to require mechanical ventilation. Multilobe involvement, history of cardiac disease, and low platelets predict poor outcomes.4 Chest crisis can result in decreased lung function,5 poor integrity of the pulmonary vessels, and a propensity to pulmonary edema. Asthma is common in patients with SCA, occurring in 17% to 53%. Asthma not only complicates the diagnosis of acute chest crisis but also increases the likelihood of chest syndrome by four- to six-fold.5
Pulmonary hypertension develops in 16% to 35% of children with SCA. By adulthood, about one-third have pulmonary hypertension, a condition that is associated with diastolic dysfunction and increased mortality. Increased intensity of the second heart sound, evidence of right ventricular enlargement, or unexplained desaturation should suggest pulmonary hypertension (see chapters 57 and 58, “Systemic Hypertension” and “Pulmonary Hypertension,” respectively, section on pulmonary hypertension). Diagnosis is made by echocardiogram and right heart cardiac catheterization demonstrating a pulmonary artery pressure above 25 mm Hg. Progressive disease can cause chest pain, dyspnea on exertion, resting hypoxia, right-sided heart failure, syncope, and pulmonary thromboembolism.
Cardiomegaly and heart murmurs are common in children with SCA. However, overt heart failure is uncommon in children, except in cases of iatrogenic volume overload associated with transfusion. Heart failure is managed by the usual methods and by correcting anemia.
Across all ages, infection is the leading cause of death in patients with SCA.6 Splenic dysfunction and inability to form immunoglobulin G antibodies to polysaccharide antigens increase susceptibility to infection, particularly with encapsulated organisms. Children age 6 months to 3 years old are at the greatest risk for sepsis, with H. influenzae and S. pneumoniae more common in the very young, and Escherichia coli and Salmonella more frequent in older children. The rate of bacteremia in children ≤3 years old has been about 8 per 100 patient-years. In children who receive pneumococcal 7-valent conjugate vaccine, invasive pneumococcal disease has decreased by as much as 93.4%.7 Table 142-1 reviews risk factors for bacteremia. Workup of potential infection in SCA patients should include a CBC, urinalysis, chest radiograph, oxygen saturation measurement, and cultures of the blood, throat, urine, or other potential source of infection.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


