INTRODUCTION
Hereditary anemias result from defects in hemoglobin production, abnormalities in red blood cell (RBC) metabolism, or changes within RBC membrane structure. Increased hemolysis occurs because the RBCs produced are either abnormal or sustain damage after release from the bone marrow, and are removed from the circulation, primarily by the spleen. Depending on the compensatory rate of production, the concentration of circulating erythrocytes may decrease, resulting in anemia.
Inherited hemoglobin disorders are comprised of two main groups: disorders with abnormal hemoglobin structure (e.g., sickle cell disease) and disorders of abnormal hemoglobin production (e.g., the thalassemias). These disorders are widely prevalent; an estimated 7% of the world’s population are carriers of an abnormal hemoglobin gene.1 These genetic abnormalities result in hemoglobin that tends to gel or crystallize, possesses abnormal oxygen-binding properties, or is readily oxidized to methemoglobin, rendering the RBC susceptible to hemolysis.
SICKLE CELL DISEASE
Sickle cell disease (SCD) is a worldwide public health problem.2 An estimated 250 million people (approximately 4.5% of the world population) are carriers of the sickle cell gene,3 with 60 million new carriers of sickle cell and 1.2 million new individuals with SCD diagnosed every year.4 SCD affects predominantly people of African Equatorial descent, although it is also found in persons of Mediterranean, Indian, and Middle Eastern origin.5,6 SCD affects approximately 70,000 people in the United States, and about 2 million Americans are sickle cell gene carriers.7
The overall life expectancy of patients in the United States with SCD is now >50 years,8 an improvement attributed to early diagnosis (antenatal and neonatal screening), parental education about complications, close monitoring in clinics and follow-up, immunizations, prophylactic penicillin to prevent pneumococcal septicemia, and increased use of drugs such as hydroxyurea.9
The normal adult RBC contains three forms of hemoglobin: HbA, HbA2, and fetal hemoglobin (HbF) (Table 236-1). Normal hemoglobin consists of a tetramer of four polypeptide chains, which are pairs of dissimilar chains (two α-globin chains and two non–α-globin chains). HbA accounts for approximately 96% to 98% of adult hemoglobin and consists of two α- and two β-globin chains. HbA2 accounts for approximately 2.0% to 3.5% of adult hemoglobin and is composed of two α- and two δ-globin chains. HbF is composed of two α- and two γ-globin chains. HbF production peaks in utero and starts declining just before birth, reaching a baseline of <1% at approximately 48 weeks of age. Because of the 120-day life span of the normal RBC, HbF is the predominant form in the circulation for approximately the first 4 months of life. The α-globin chains are coded by four genes, two each on chromosomes 16, whereas the β-, γ-, and δ-globin chains are coded by two genes, one each on chromosomes 11.
| Syndrome | Types of Hemoglobin (Hb) Present | Percentage within the Red Blood Cell | Hemoglobin Tetramer Composition (globin chains) |
|---|---|---|---|
| Normal adults | HbA | 96–98 | Two α-chains and two β-chains |
| HbA2 | 3.0–3.5 | Two α-chains and two δ-chains | |
| HbF | 0.5–0.8 | Two α-chains and two γ-chains | |
| Sickle cell trait (heterozygous) | HbA | 60–65 | Two α-chains and two β-chains |
| HbAS | 35–40 | Two α-chains, one normal β-chain, and one sickle β-chain | |
| HbF | 0.5–0.8 | Two α-chains and two γ-chains | |
| Sickle cell disease (homozygous) | HbS | 80–90 | Two α-chains and two sickle β-chains |
| HbA2 | 2–4 | Two α-chains and two γ-chains | |
| HbF | 2–20 | Two α-chains and two γ-chains |
SCD is caused by the presence of an abnormal β-globin chain.10 The specific mutation is an adenine-to-thymine substitution in the codon that results in the hydrophobic amino acid, valine, replacing the hydrophilic amino acid glutamic acid in the sixth amino acid position of the β-globin chain. As a result, under deoxygenated conditions, valine becomes buried in a hydrophobic pocket on an adjacent chain. The globin chains then join together, and HbS polymerizes, deforming the RBC and producing the characteristic sickled appearance (Figure 236-1).
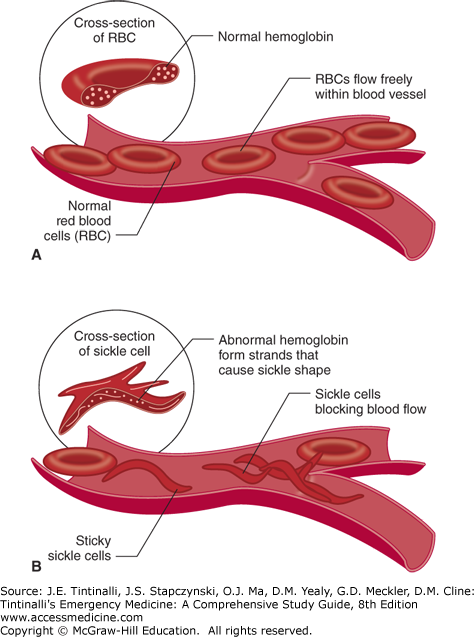
The distorted sickle cell results in premature RBC destruction (life span of about 20 days) and also increases the viscosity of blood, leading to obstruction within the microvasculature (Figure 236-1). The overall effect is chronic, ongoing hemolysis and episodic periods of vascular occlusion, resulting in tissue ischemia affecting many organ systems. Although the sickle cell gene results from a single base substitution, there is tremendous variability in the phenotypic expression of this mutation.6,11,12
SCD is seen in patients who are homozygous for the sickle gene or who are double heterozygous with another hemoglobin variant (e.g., HbSC). Sickle cell anemia refers to homozygosity of the mutation, also termed HbSS or SS disease.
People with sickle cell trait (HbAS, heterozygous with one gene for the sickle mutation) have a normal life span and usually are asymptomatic, although complications occur, grouped as definite, probable, or possible.13 Definite associations include renal medullary carcinoma, hematuria and renal papillary necrosis, hyposthenuria, splenic infarcts, and exercise-related deaths. Probable associations include venous thromboembolic events, pregnancy-related complications, and complicated hyphema. Possible associations include retinopathy, acute chest syndrome, and asymptomatic bacteruria.13 Many of the associations are seen only under conditions of severe tissue hypoxia, acidosis, dehydration, or hypothermia.
Polymerization of deoxygenated HbS, deforming the red cell into a sickled shape that causes microvascular sludging and obstruction (vaso-occlusion), is the major pathophysiologic process in SCD.10 Vascular obstruction worsens hypoxia and causes acidosis in the microcirculation that contributes to further sickling. The sickling process is initially reversible when the HbS is reoxygenated, but with repeated episodes of sickling, the red cell membrane is permanently damaged, and the cell remains irreversibly sickled. Anywhere from 5% to 50% of the circulating erythrocytes in a patient with SCD can be irreversibly sickled cells. The sickling process is inhibited by the presence of HbF and other hemoglobin variants.14 Structural and antigenic changes on the RBC membrane cause an abnormal tendency for the erythrocyte to catalyze plasma coagulation factors and adhere to vascular endothelium in SCD.
Because newborn screening occurs in the United States and in most developed countries, the majority of patients who present to the ED will already know that they have the sickle cell trait or disease. Although SCD can produce a wide spectrum of manifestations, vaso-occlusive crisis accounts for the majority of ED presentations as the sickled RBCs restrict the blood flow to various organs, thus causing ischemic pain and organ damage.10,15,16 Patients may also present to the ED with life-threatening complications such as stroke, aplastic crisis, acute chest syndrome, and sepsis17,18 (Table 236-2).
| Neurologic | Ischemic or hemorrhagic stroke Cerebral aneurysm Chronic pain syndrome Neuropathic pain syndrome |
| Eye | Retinopathy |
| Pulmonary | Acute chest syndrome Pulmonary hypertension |
| Cardiac | Cardiomegaly |
| Abdominal, GI | Mesenteric ischemia Hepatic infarction Cholelithiasis Intrahepatic cholestasis Splenic sequestration |
| Renal, GU | Hematuria Renal infarction Papillary necrosis Renal failure Priapism |
| Musculoskeletal | Acute vaso-occlusive pain crisis; bone, joint, and muscle pain Osteomyelitis Avascular necrosis of femoral head |
| Dermatologic | Leg ulcers |
| Infection | Osteomyelitis Pneumonia Urinary tract infection |
| Hematologic | Chronic hemolysis Acute hemolytic crisis Aplastic crisis Hypercoagulability: pulmonary embolism and venous thrombosis |
Acute vaso-occlusive pain crisis is a common problem in patients with SCD.10,16 The initiating event may not be identifiable, but stressors such as infection, cold, dehydration, and altitude have been implicated. As a result of intravascular sickling and small-vessel occlusion, infarction of bone, viscera, and soft tissue occurs. This is manifested as diffuse bone, muscle, and joint pain and, in some cases, symptoms related to a specific affected organ. Initial management of these patients includes aggressive pain management, appropriate hydration, and an assessment for a treatable cause of the current crisis as well as a search for additional complications (Table 236-3).1,18,19,20
| History | Duration and location of pain History of fever Presence of focal swelling or redness Precipitation factors for acute episode Medications taken for pain relief |
| Physical examination | Assess degree of pain Inspect sites of pain, looking for swelling, warmth, redness General: respiratory distress, pallor, hydration, jaundice, rash Vital signs: especially temperature, pulse oximetry Respiratory: lung sounds Heart: cardiomegaly and systolic murmur common with chronic anemia Abdomen: tenderness, organomegaly |
| Ancillary tests | Obtain if moderate to severe pain, focal pathology is present, or pain is atypical for acute episode CBC, leukocyte differential, reticulocyte count, urinalysis Chest radiograph, if signs of lower respiratory tract pathology Blood cultures and additional blood tests: as indicated by clinical condition |
| General management | Bed rest, provide warmth and a calm, relaxing atmosphere Distractions where appropriate: television, music, etc. Oral fluids: typically about 3 L/d IV fluids to correct dehydration or if reluctant to drink or vomiting is present Oxygen: not routinely required unless hypoxemia is present Encourage deep breathing, incentive spirometry |
| Pain management | Use analgesics appropriate to degree of pain Acetaminophen for mild pain Nonsteroidal anti-inflammatory drug for mild to moderate pain (avoid if renal insufficiency is present) Opioids for moderate to severe pain; typical initial doses include:
Reassess response in 15–30 min; may repeat with one fourth to one half of the initial dose |
| Consider adjuvant therapy | Medication to prevent constipation Antiemetic Anxiolytic Tinzaparin |
| Disposition and follow-up | Consider admission to the hospital if:
|
Consider discharge if:
|
The CBC and reticulocyte count assess the degree of anemia and whether the marrow is still producing red cells. If the reticulocyte count is not available, the presence of polychromasia in the peripheral blood smear can be used to provide evidence of continued RBC production. It is common for SCD patients to have a low-grade temperature as well as a modestly elevated leukocyte count during a painful crisis, potentially confounding detection of an infection. A leukocyte count >20,000/mm3 (> 20 × 109/L) with an increased number of bands is not typical for sickle crisis alone; this combination suggests a potential infection. Mild elevations in serum bilirubin and lactate dehydrogenase levels are common due to chronic hemolysis.
Supplemental oxygen has no proven benefit, unless the patient is systemically hypoxemic.18,20 SCD patients during a painful crisis may be hypovolemic due to their disease (deficient renal concentrating ability) or crisis (anorexia, vomiting, or fever), so PO or IV rehydration may be necessary. No hydration regimen has proven capable of shortening the duration or severity of a painful sickle cell attack.21 After rehydration, maintenance PO or IV (5% dextrose in 0.5 normal saline) should be provided.
Treatment of acute, moderate to severe pain usually requires opioids,1,10,16,18,19,20 usually parenteral, but sustained-release oral morphine may be as effective as parenteral morphine.22 Potent opioids, such as morphine or hydromorphone, are recommended, but meperidine, with the potential for neurotoxicity (seizures) from accumulation of the metabolite normeperidine, is not.20 Some patients, because of prior opioid treatment, may be very tolerant, and large doses may be required to achieve adequate analgesia. Regular doses of analgesics for a few hours to several days typically are required. Patient-controlled analgesia has been used in selected patients. Nonsteroidal anti-inflammatory drugs can be used for their probable additive effect in pain management of sickle cell crisis.18 Low-dose ketamine by continuous IV infusion is a potential adjuvant analgesic in acute pain crisis.23
Tinzaparin, a low-molecular-weight heparin that has antithrombotic, anti-inflammatory, and endovascular effects may be considered in protracted vaso-occlusive crises1,24; the recommended dose is 175 IU/kg SC daily for 2 to 7 days.
A recommended practice is to develop an individualized assessment and treatment protocol for specific patients who frequently present to the ED with painful crises.20
RBC transfusion to reduce the concentration of HbS-containing erythrocytes does not shorten the duration or reduce the risk of complications of routine acute vaso-occlusive painful crisis.25 Furthermore, transfusion involves significant expense, the risk of bloodborne disease transmission, and the potential for iron overload and exposes the patient to the minor RBC antigens, with the potential to induce antibodies that prevent or complicate future transfusions (see chapter 238, “Transfusion Therapy”).26,27 Transfusion for sickle cell crisis or complications is reserved for specific indications such as aplastic crisis, pregnancy, stroke, respiratory failure, and general surgery.10,28,29 Transfusion protocols in SCD are categorized as aggressive (decreasing HbS to <30%) and conservative (increasing hemoglobin to >10 grams/dL or 100 g/L); for most indications, there is no clinical difference between aggressive or conservative protocols.29
Hydroxyurea (hydroxycarbamide) successfully reduces the frequency and severity of painful crises in SCD.8,30,31,32,33 Hydroxyurea blocks the synthesis of DNA and impairs cell division. It also increases the production of HbF to levels that are protective and preventive of RBC sickling. Hydroxyurea is indicated for young adults who have three or more hospitalizations for vaso-occlusive crisis in the preceding 12 months.8,31 Daily prophylactic penicillin V reduces the incidence of infections and reduces mortality from sepsis in children but with little benefit in adults.34
Bone pain is common during a sickle cell crisis and is usually located in the back and the extremities.35 The pain will be diffuse, without focal signs of inflammation; therefore redness, warmth, or swelling suggests infection such as cellulitis or osteomyelitis. Localized hip pain with difficulty ambulating suggests aseptic necrosis of the femoral head; approximately 30% of SCD patients develop femoral head damage by age 30 years old. Joint effusions are occasionally seen in painful sickle cell crisis, and arthrocentesis is often necessary to differentiate this complication from infection. Plain radiographs may show evidence of aseptic necrosis or osteomyelitis, whereas bone infarcts usually are not visible on radiographs. A radionuclide bone scan or MRI may be necessary to differentiate infection from infarction.
Acute chest syndrome is defined as a new infiltrate on chest radiograph in association with one other new sign or symptom: fever >38.5°C (101.3°F), cough, wheezing, tachypnea, or chest pains.36,37,38 In adults, acute chest syndrome occurs most commonly 1 to 3 days after hospitalization for an acute pain crisis. Acute chest syndrome is the leading cause of death in patients with SCD in the United States.39 Although this phenomenon occurs most often as a single episode, some patients may experience multiple attacks, resulting in chronic lung disease.40 Also, symptoms may vary with repeated episodes.
Acute chest syndrome has multiple potential etiologies; most commonly, the syndrome is precipitated by pulmonary infections, fat emboli, and rib infarction.37,38,40 Acute chest syndrome may also result from iatrogenic causes, such as aggressive hydration for sickle cell painful crisis producing pulmonary edema and opioid use depressing inspiratory effort and promoting atelectasis.
Infectious pathogens were identified in patients admitted to the hospital with acute chest syndrome in a national study.36 The two most common organisms identified were the atypical pneumonia pathogens Chlamydia pneumoniae and Mycoplasma pneumoniae.36 Other organisms associated with acute chest syndrome include Staphylococcus aureus, Haemophilus influenzae, Klebsiella pneumoniae, adenoviruses, influenza viruses, parainfluenza viruses, respiratory syncytial viruses, cytomegalovirus, and parvovirus B19.36 Traditionally, Streptococcus pneumoniae was considered the most frequent cause of pulmonary infection in SCD patients, but this pathogen is now rare in cases of acute chest syndrome, presumably due to the use of pneumococcal immunization and prophylactic penicillin therapy.
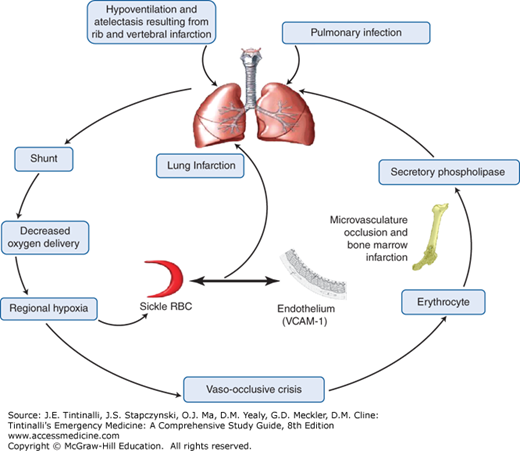
Abundant evidence for fat embolism as a possible cause for acute chest syndrome includes bony slivers and marrow fat found in the pulmonary vasculature at autopsy, fat droplets within endothelial cells identified in lung biopsy specimens collected via bronchoscopy in living patients, and elevated serum levels of free fatty acids and circulating secretory phospholipase A2, a potent inflammatory mediator originating from the bone marrow.41 It is postulated that reduced blood flow to the bone marrow during a vaso-occlusive crisis results in ischemia and necrosis, so that pieces of necrotic marrow embolize and become lodged in the pulmonary vasculature, playing a role in the pathophysiology of acute chest syndrome.
The combination of both ischemia and hypoxia in the pulmonary circulation promotes the production of free radicals, which further results in the upregulation of endothelial adhesion molecules such as vascular cell adhesion molecule-1. This activation causes binding of sickled RBC and leukocytes to the endothelium, which further increases the cycle of vaso-occlusion. The acute chest syndrome is the final result of several pathogenic processes exerting their effects individually and collectively in a vicious cycle, creating regional hypoxia, acidosis, and lung injury (Figure 236-2).
Respiratory symptoms are usually present, including cough, shortness of breath, chest pain, and fever, although the clinical presentation may vary.37,38 Chest radiographs are important to identify the presence of a new patchy infiltrate; any lobe can be affected. The radiographic changes often lag behind the clinical features, so the radiograph may be initially normal, and the clinical severity of disease and the extent of hypoxia may not correlate with radiographs.
Acute chest syndrome is treated with supportive care (oxygen, analgesics, hydration), antibiotics, and exchange transfusion (Table 236-4).37,38,40 Oxygen should be administered, especially if hypoxemia is demonstrated. Pain control initially requires high doses of parenteral opioids but requires careful monitoring to prevent both oversedation and hypoventilation. Dehydration results in increased plasma osmolarity and intracellular dehydration of RBCs, resulting in the further propagation of the sickling phenomenon. Supportive IV fluid therapy (up to 1.5 times maintenance) in the form of hypotonic solution facilitates free water passage into the relatively hypertonic red cells so that the resultant osmotic swelling decreases the mean corpuscular hemoglobin concentration and reduces the tendency for sickling.
| History | Major presenting symptoms: dyspnea, fever, cough Accompanying chest, rib, bone, or joint pain Assess degree or severity of pain Recent or previous sepsis, infection, pneumonia, or hospitalization Prior history of acute chest syndrome, especially if required intubation and ventilatory support Potentially infectious contacts Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|




