Shock, Ischemia, and Reperfusion Injury
Basilia Zingarelli
KEY POINTS
Ischemia and Reperfusion
 Ischemia results from a dramatic reduction of oxygen supply to tissues and/or organs and rapidly results in cell metabolic derangement, molecular alterations, and dysfunction sequelae.
Ischemia results from a dramatic reduction of oxygen supply to tissues and/or organs and rapidly results in cell metabolic derangement, molecular alterations, and dysfunction sequelae. Timely reperfusion of the ischemic tissue is the mandatory treatment of patients with clinical conditions of ischemia. However, reperfusion initiates a cascade of events that causes additional cell injury, a phenomenon called reperfusion injury.
Timely reperfusion of the ischemic tissue is the mandatory treatment of patients with clinical conditions of ischemia. However, reperfusion initiates a cascade of events that causes additional cell injury, a phenomenon called reperfusion injury.Oxidative and Nitrosative Stress
 The production of excessive quantities of reactive oxygen species (ROS) and reactive nitrogen species (RNS) is an important mechanism of reperfusion injury. These reactive species mediate tissue injury by two main mechanisms: directly, by inducing damage of important cellular macromolecules, and indirectly, by activating signal transduction pathways.
The production of excessive quantities of reactive oxygen species (ROS) and reactive nitrogen species (RNS) is an important mechanism of reperfusion injury. These reactive species mediate tissue injury by two main mechanisms: directly, by inducing damage of important cellular macromolecules, and indirectly, by activating signal transduction pathways.
Activation of Signaling Pathways
 Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) contribute to activation of inflammatory signaling pathway by recruiting toll-like receptors (TLRs) in the host cells.
Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) contribute to activation of inflammatory signaling pathway by recruiting toll-like receptors (TLRs) in the host cells. ROS and RNS initiate signaling cascades that result in a prompt production of inflammatory mediators, such as cytokines, adhesion molecules, chemokines, and metabolic enzymes, thus determining the functional outcome of the cell in response to ischemia.
ROS and RNS initiate signaling cascades that result in a prompt production of inflammatory mediators, such as cytokines, adhesion molecules, chemokines, and metabolic enzymes, thus determining the functional outcome of the cell in response to ischemia. The complement system is activated in an attempt to induce lysis of foreign organisms and to promote phagocytic clearance of immune complexes and the recruitment of inflammatory cells at the site of injury. However, this system can amplify endothelial dysfunction and the parenchymal damage during reperfusion.
The complement system is activated in an attempt to induce lysis of foreign organisms and to promote phagocytic clearance of immune complexes and the recruitment of inflammatory cells at the site of injury. However, this system can amplify endothelial dysfunction and the parenchymal damage during reperfusion.Endothelial Dysfunction and Organ Injury
 Endothelial dysfunction is triggered by the endothelial generation of ROS and RNS and is characterized by the development of a procoagulant phenotype, loss of vascular responsiveness to vasodilator and vasoconstrictor agents, increased vascular permeability, and adhesiveness to platelets and leukocytes.
Endothelial dysfunction is triggered by the endothelial generation of ROS and RNS and is characterized by the development of a procoagulant phenotype, loss of vascular responsiveness to vasodilator and vasoconstrictor agents, increased vascular permeability, and adhesiveness to platelets and leukocytes. In the early phase of shock and reperfusion, leukocytes are present mainly in the intravascular space (attached to the endothelium), and they play an important role in endothelial injury. At later stages, leukocytes migrate into the tissues and exacerbate parenchymal damage.
In the early phase of shock and reperfusion, leukocytes are present mainly in the intravascular space (attached to the endothelium), and they play an important role in endothelial injury. At later stages, leukocytes migrate into the tissues and exacerbate parenchymal damage. The production of ROS and RNS, endothelial injury, activation of the complement system, release of neutrophilattractive factors, and leukocyte infiltration (leading to further production of reactive species and proteolytic enzymes) constitute a vicious cycle, which is ultimately responsible for organ injury.
The production of ROS and RNS, endothelial injury, activation of the complement system, release of neutrophilattractive factors, and leukocyte infiltration (leading to further production of reactive species and proteolytic enzymes) constitute a vicious cycle, which is ultimately responsible for organ injury.Future Prospects
 Current experimental research has raised the exciting prospect that pharmacologic intervention aimed at interrupting the various levels of the ischemia-reperfusion injury cycle may ameliorate cell dysfunction and prevent death. However, large clinical trials are necessary to determine whether these novel therapeutic interventions may be beneficial to the patient.
Current experimental research has raised the exciting prospect that pharmacologic intervention aimed at interrupting the various levels of the ischemia-reperfusion injury cycle may ameliorate cell dysfunction and prevent death. However, large clinical trials are necessary to determine whether these novel therapeutic interventions may be beneficial to the patient.Ischemia and reperfusion injury is a critical pathophysiologic sequence common to several clinical conditions, including myocardial infarction, intestinal ischemia, stroke, cardiopulmonary bypass, solid-organ transplantation, soft-tissue flaps, and extremity reimplantation. It also occurs as a consequence of collapse of systemic circulation, as in shock with low cardiac output, which results in inadequate tissue perfusion (ischemia) and typically requires cardiovascular resuscitation (reperfusion).
Ischemia can result from a dramatic reduction of oxygen supply in the whole organism or in defined tissue regions and rapidly results in cellular metabolic derangements, molecular  alterations, and dysfunction sequelae. Occasionally, ischemia may occur (despite adequate oxygen delivery) from either increased metabolic requirements or impaired oxygen utilization at the cellular level. If not reversed within a short period, the altered cellular metabolism progresses to complete depletion of
alterations, and dysfunction sequelae. Occasionally, ischemia may occur (despite adequate oxygen delivery) from either increased metabolic requirements or impaired oxygen utilization at the cellular level. If not reversed within a short period, the altered cellular metabolism progresses to complete depletion of
the energetic pools, accumulation of waste and toxic products, and eventually, to cell death.
alterations, and dysfunction sequelae. Occasionally, ischemia may occur (despite adequate oxygen delivery) from either increased metabolic requirements or impaired oxygen utilization at the cellular level. If not reversed within a short period, the altered cellular metabolism progresses to complete depletion of the energetic pools, accumulation of waste and toxic products, and eventually, to cell death.
The main therapeutic intervention in clinical conditions of ischemia requires the restoration of blood flow (reperfusion) and/or recovery of the normal oxygen levels (reoxygenation) by providing circulatory and respiratory support. Once perfusion is reestablished, tissue ischemia is generally reversed. However, restoration of blood flow initiates a paradoxical injury process  known as reperfusion injury, which is characterized by dysregulation of the inflammatory and innate immune responses and may lead to cellular death and further organ dysfunction. The intense inflammation triggered by ischemia and reperfusion injury may also precipitate inflammatory damage in organs not involved in the initial ischemic insult. This condition is termed multiple organ dysfunction syndrome (MODS) (see Chapter 23) and is the leading cause of death in ICUs.
known as reperfusion injury, which is characterized by dysregulation of the inflammatory and innate immune responses and may lead to cellular death and further organ dysfunction. The intense inflammation triggered by ischemia and reperfusion injury may also precipitate inflammatory damage in organs not involved in the initial ischemic insult. This condition is termed multiple organ dysfunction syndrome (MODS) (see Chapter 23) and is the leading cause of death in ICUs.
known as reperfusion injury, which is characterized by dysregulation of the inflammatory and innate immune responses and may lead to cellular death and further organ dysfunction. The intense inflammation triggered by ischemia and reperfusion injury may also precipitate inflammatory damage in organs not involved in the initial ischemic insult. This condition is termed multiple organ dysfunction syndrome (MODS) (see Chapter 23) and is the leading cause of death in ICUs.The biochemical and molecular changes of ischemia and reperfusion are complex; these include (a) the formation of toxic inflammatory mediators, reactive oxygen species (ROS) and reactive nitrogen species (RNS) by inflammatory and parenchymal cells, (b) the disturbances of the microcirculation and endothelium barrier, and (c) the alteration of the coagulation and complement cascades. To counteract this deleterious inflammatory response, the tissues also attempt to adapt within the altered metabolic environment by deploying cellular mechanisms of defense (see Chapter 20).
OXYGEN DELIVERY AND ACUTE CELLULAR OXYGEN DEFICIENCY
According to the Fick equation, oxygen delivery (DO2) depends on both the arterial oxygen content (CaO2) and the cardiac output (CO). The CO depends on the heart rate and the stroke volume and the stroke volume is determined by myocardial contractility and ventricular preload and afterload. In infants and children, CO depends more on heart rate than on stroke volume because of an incomplete development of myocardial contractile mass. Compensatory changes in the local vasomotor tone modify the systemic vascular resistance and adjust CO to maintain tissue perfusion and meet metabolic demands (Fig. 21.1).
Therefore, acute cellular oxygen deficiency may result from a reduction of DO2 due to local tissue ischemia within the perfusion territory of an occluded artery (myocardial or splanchnic infarction, cerebral ischemia, solid-organ transplantation, soft-tissue and limb reimplantation). Conversely, decreased CO may cause global tissue ischemia observed in conditions of cardiogenic shock. However, in some clinical conditions, inadequate tissue energetic metabolism may derive from an increase of total body oxygen consumption (VO2), despite a normal DO2. For example, during septic shock tissue oxygenation may be inadequate even in the presence of even increased blood flow due to a major increase in metabolic demand and impaired oxygen extraction.
DEFINITION AND CLASSIFICATION OF SHOCK
Shock is defined as a state of acute circulatory dysfunction that results in a failure to deliver sufficient oxygen and other nutrients to meet the metabolic demands of the tissues (1). Shock is a progressive process whose three stages are characterized by the causative factors, the cellular compensatory responses, and the ischemia and reperfusion injury. In the early or compensated stage, a number of neurohormonal compensatory physiologic mechanisms act to maintain blood pressure and preserve adequate tissue perfusion. At this stage, shock may be reversible, even without therapeutic intervention. When the compensatory mechanisms fail, shock progresses to an uncompensated stage that requires, and still responds to, therapeutic interventions. In the irreversible stage, shock progresses to a remarkable degree of organ and tissue injury, which is unresponsive to conventional therapy and leads to the death of the patient.
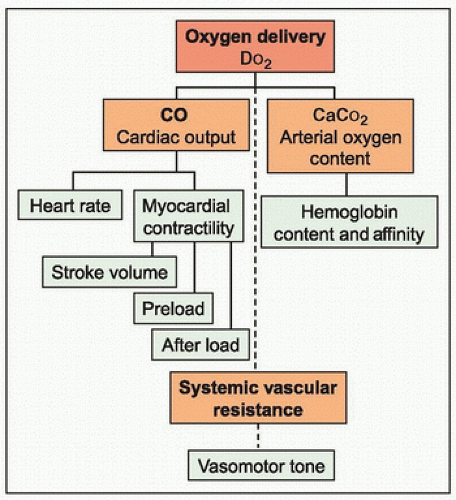 FIGURE 21.1. Variables of oxygen delivery. Oxygen delivery (DO2) depends on cardiac output (CO) and arterial oxygen content (CaO2). CO depends on heart rate and stroke volume, which is determined by myocardial contractility and the ventricular preload and afterload. CaO2 depends on hemoglobin content, affinity, and saturation. Modification of the systemic vascular resistance contributes to maintenance of tissue perfusion through changes in the local vasomotor tone and affects CO by altering afterload. |
Five main types of shock are described (Table 21.1). (a) Hypovolemic shock is the most common type of shock in children and is caused by a reduction in the volume of blood by hemorrhage or dehydration (especially in young children). (b) Cardiogenic shock is caused by a decline in CO secondary to myocardial damage and/or dysfunction. This type of shock is less frequent in children but may be seen in neonates with congenital heart disease, in older patients immediately after repair of congenital heart defects, and in patients with myocarditis, thyrotoxicosis, and pheochromocytoma. Septic shock may be included in cardiogenic shock because of the occurrence of myocardial depression induced by inflammatory mediators. (c) Distributive shock results from abnormalities in flow distribution secondary to impaired vasomotor tone, despite a normal or high CO. The most common cause of distributive shock in children is sepsis, especially in the early stage. Other types of distributive shock are anaphylactic shock from systemic allergic reactions and neurologic shock from spinal cord injuries. Drug intoxication by barbiturates, phenothiazines, and antihypertensives may also cause maldistribution of blood flow. (d) Obstructive shock is uncommon in children and is caused by a mechanical obstruction that impairs CO. The common causes include pericardial tamponade, congenital heart lesions characterized by left ventricular outflow obstruction (e.g., critical aortic stenosis, hypertrophic cardiomyopathy), tension pneumothorax and, less frequently, pulmonary embolism. (e) Dissociative shock is also uncommon
in children and results from clinical conditions associated with inadequate tissue oxygenation secondary to abnormal affinity of hemoglobin for oxygen, such as methemoglobinemia and carbon monoxide poisoning.
in children and results from clinical conditions associated with inadequate tissue oxygenation secondary to abnormal affinity of hemoglobin for oxygen, such as methemoglobinemia and carbon monoxide poisoning.
TABLE 21.1 CLASSIFICATION OF SHOCK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The specific pattern of response and related pathophysiology, clinical manifestations, and treatments vary with the etiology of shock (see Chapter 28).
CELLULAR AND MOLECULAR MECHANISMS OF INJURY
Metabolic Derangement and Energy Failure during Ischemia
The cells of all tissues undergo metabolic changes when deprived of oxygen and other nutrients. The quantitative and kinetic features of these metabolic responses are different for each specialized cell type and depend on the function and structure of the cell. Brain and myocardium are the most vulnerable tissues to acute oxygen deficiency, and even a short period of ischemia can lead to deleterious and irreversible effects.
The cause of shock or ischemia also has profound effects on many fundamental aspects of cell metabolism and function. The pathophysiologic effects of cardiogenic and hypovolemic shock are related predominantly to the acute decline in DO2, whereas the pathophysiologic effects of sepsis result largely from the overwhelming production of inflammatory mediators. In shock associated with sepsis, in fact, bacterial products interact directly with several cell types, including macrophages, neutrophils, monocytes, endothelial cells, and myocytes to activate the innate immune response and to stimulate the production of numerous inflammatory toxic mediators. The excessive inflammatory response is then responsible for hemodynamic compromise, widespread tissue ischemia, and reperfusion injury (Fig. 21.2).
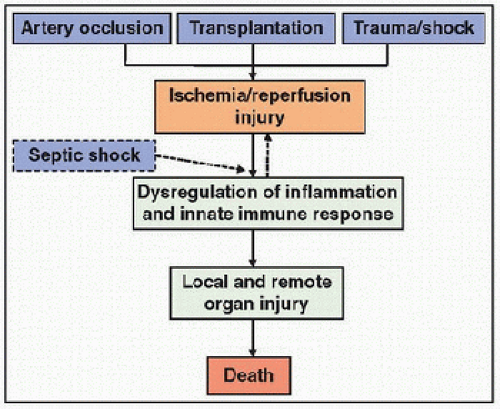 FIGURE 21.2. Kinetics of pathophysiologic events of shock and reperfusion. The pathophysiologic effects of acute local ischemia (after artery occlusion or organ transplantation) or acute global ischemia (after trauma and severe hemorrhage) are related predominantly to the acute deficiency of oxygen delivery and the subsequent reperfusion injury at the time of reoxygenation. In septic shock, an overwhelming inflammatory response is first activated by bacterial products and precedes the hemodynamic compromise and widespread tissue ischemia and reperfusion injury. |
Independent of the cause of ischemia or cell specificity, a time-dependent cascade of metabolic responses is shared among all cell types during ischemia (Fig. 21.3). Under normal conditions, glucose is converted into pyruvate via the glycolytic pathway. Pyruvate in turn is taken up by mitochondria and enters the Krebs cycle, where it is used in the production of adenosine triphosphate (ATP) via the electron transport chain (a process referred to as aerobic metabolism). After only a few seconds of ischemia, oxidative phosphorylation and mitochondrial ATP production are seriously compromised. The cell shifts from oxidative metabolism to an inefficient anaerobic glycolysis, where pyruvate remains in the cytoplasm and is converted into lactate, a process that produces only 2 moles of ATP per 1 mole of glucose, instead of the 38 moles of ATP normally produced during aerobic metabolism. Therefore, levels of ATP and other high-energy phosphates—mainly creatine phosphate—decline rapidly, and the breakdown products of adenine nucleotides, such as inorganic phosphate and adenosine, accumulate. This collapse of high-energy phosphate compounds impairs the energy-dependent cell processes, especially membrane ion pumps (Na+/H+ exchanger). DNA and protein synthesis are also suppressed, although some specific proteins, for example, heat shock protein 70 (Hsp70) and protein kinase C (PKC), may be induced in an attempt by the cell to adapt to the hypoxic environment (see Chapter 20).
Increased anaerobic glycolysis also leads to the accumulation of hydrogen ions, lactate, and intracellular acidosis. The elevated lactate levels can be measured in the serum and used as a biomarker of the ensuing shock state. Membrane depolarization and failure of ATP-dependent ion pumps lead to an efflux of potassium and influx of sodium and calcium. The increase of intracellular sodium concentration is accompanied by water influx into the cell, leading to swelling of the
cytoplasm and organelles such as mitochondria and endoplasmic reticulum. These abnormalities become rapidly severe and may reduce cellular functions within minutes. In the heart, this metabolic derangement causes a reduction of the contractile force generated by actin-myosin cross-bridging interactions. In the gut, the result is an altered intestinal absorptive function that is associated with translocation of bacteria from the intestine to the lymphatic vessels and blood stream. At this point, cell injury is reversible if oxygen supply is restored.
cytoplasm and organelles such as mitochondria and endoplasmic reticulum. These abnormalities become rapidly severe and may reduce cellular functions within minutes. In the heart, this metabolic derangement causes a reduction of the contractile force generated by actin-myosin cross-bridging interactions. In the gut, the result is an altered intestinal absorptive function that is associated with translocation of bacteria from the intestine to the lymphatic vessels and blood stream. At this point, cell injury is reversible if oxygen supply is restored.
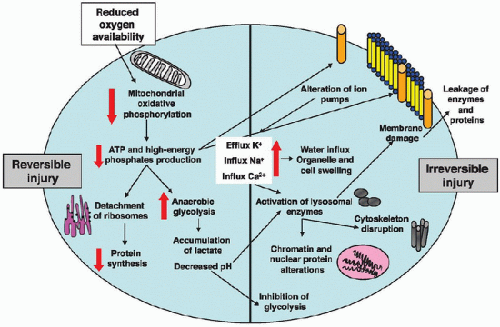 FIGURE 21.3. Metabolic and cellular derangement during ischemia. Reduction in oxygen availability compromises mitochondrial oxidative phosphorylation. Levels of ATP and other high-energy phosphates rapidly decline. The cell shifts from oxidative metabolism to an inefficient anaerobic glycolysis, producing less ATP. This energy failure slows the rate of protein synthesis and impairs the energy-dependent ion pumps, leading to an increase of intracellular levels of sodium (Na+) and calcium (Ca2+), an influx of water, and a decrease of intracellular levels of potassium (K+). Water influx into the cell leads to swelling of the cytoplasm and organelles, such as mitochondria and endoplasmic reticulum, further impairing mitochondrial oxidative phosphorylation and protein synthesis. Accumulation of hydrogen ions and lactate results in intracellular acidosis and inhibition of glycolysis. At this time, if oxygen supply is not restored, cell injury becomes irreversible. Increase of intracellular Ca2+ causes activation of lysosomal enzymes and disrupts mitochondria, further uncoupling oxidative phosphorylation. Activated proteases also cleave cytoskeletal filaments and impair membrane permeability. Finally, physical disruption of the cell membrane occurs and is associated with leakage of cellular enzymes and proteins into the plasma. |
However, if ischemia persists, cell injury may become irreversible. The calcium concentration in the cytosol and mitochondria also increases due to changes in the cell ATPasedependent ion transport systems. Mitochondria show amorphous matrix densities and granular dense bodies of calcium phosphate, which are considered the earliest sign of irreversible ischemic cell injury. The excessive energy demand and the resulting decrease in adenine nucleotide concentration represent crucial factors in the ensuing irreversible damage of cells.
This altered metabolic milieu, with a sustained increase in cytosolic and mitochondrial calcium and low internal pH (pHi), causes the activation of a number of enzymes (proteases, phospholipases, ATPases), which disrupt mitochondrial and lysosomal membranes, further uncoupling mitochondrial oxidative phosphorylation and promoting the release of other acid hydrolases. Activated proteases also cleave cytoskeletal filaments and impair their anchoring and stabilizing function. These changes collectively lead to a progressive increase in membrane permeability, severe derangements of intracellular electrolytes, and ATP exhaustion (2).
The terminal event after prolonged ischemia is cell death, which occurs mainly by necrosis and is associated with physical disruption of the cell membrane and leakage of cellular enzymes and proteins across the cell membrane and into the plasma. This latter phenomenon may provide important diagnostic tools of cell damage, such as the serum increase of creatine kinase and troponin levels for the diagnosis of myocardial infarction.
Reperfusion Injury
Rapid and sustained restoration of oxygen and nutrient supply to the ischemic tissue is the major goal in the treatment of patients with clinical conditions of global or local ischemia. This
therapeutic intervention has the potential to relieve ischemia and can result in the recovery of cell function and prevention of cell death. Upon reperfusion, if the cell injury is still reversible, electron transfer and ATP synthesis restart, the pHi is restored, and other metabolic processes are reestablished. The intensity and duration of ischemia are the main factors that influence recovery: earlier reperfusion and reoxygenation are associated with a more favorable clinical outcome.
therapeutic intervention has the potential to relieve ischemia and can result in the recovery of cell function and prevention of cell death. Upon reperfusion, if the cell injury is still reversible, electron transfer and ATP synthesis restart, the pHi is restored, and other metabolic processes are reestablished. The intensity and duration of ischemia are the main factors that influence recovery: earlier reperfusion and reoxygenation are associated with a more favorable clinical outcome.
Paradoxically, reoxygenation or restoration of blood flow initiates a cascade of events that results to even further cell injury, known as reperfusion injury. The underlying pathophysiologic mechanisms of this phenomenon are complex and involve inflammatory reactions as well as activation of the innate immune system. Major contributing events to reperfusion injury are oxidative and nitrosative stress, endothelial dysfunction, microvascular injury, neutrophil activation, and complement system activation.
Oxidative and Nitrosative Stress
The production of excessive quantities of ROS and RNS is  an important mechanism of reperfusion injury. These reactive species include highly unstable oxygen and nitrogen molecules, such as superoxide (O2–), hydrogen peroxide (H2O2), oxidized lipoproteins, lipid peroxides, nitric oxide (NO), and peroxynitrite (ONOO) (3). ROS and RNS can be formed by several mechanisms; they can be produced by a reduction of molecular oxygen in altered mitochondria; by enzymes such as xanthine oxidase (XO), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (also called Nox), cytochrome P450, nitric oxide synthase (NOS), and cyclooxygenase (COX); and by auto-oxidation of catecholamines. The source of reactive species varies from cell to cell and from stage to stage during the ischemia and reperfusion event. For example, in myocytes, but to a much lesser extent than in other cell types, the production of oxyradicals starts early in the ischemic period in altered mitochondria and is further enhanced during reperfusion. At reperfusion, a massive activation of XO is a major source of ROS in endothelial cells, and COX may contribute to this oxidative stress. Cytochrome P450 accounts for the production of ROS in lung epithelial cells, and NADPH oxidase accounts for ROS production in neutrophils that have infiltrated the affected tissues. The production of potent RNS appears to be more ubiquitous because a number of inflammatory mediators may lead to the induction of NOS in several cell types. In addition, the antioxidant mechanisms of the cell may be compromised and may further favor the accumulation of reactive species.
an important mechanism of reperfusion injury. These reactive species include highly unstable oxygen and nitrogen molecules, such as superoxide (O2–), hydrogen peroxide (H2O2), oxidized lipoproteins, lipid peroxides, nitric oxide (NO), and peroxynitrite (ONOO) (3). ROS and RNS can be formed by several mechanisms; they can be produced by a reduction of molecular oxygen in altered mitochondria; by enzymes such as xanthine oxidase (XO), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (also called Nox), cytochrome P450, nitric oxide synthase (NOS), and cyclooxygenase (COX); and by auto-oxidation of catecholamines. The source of reactive species varies from cell to cell and from stage to stage during the ischemia and reperfusion event. For example, in myocytes, but to a much lesser extent than in other cell types, the production of oxyradicals starts early in the ischemic period in altered mitochondria and is further enhanced during reperfusion. At reperfusion, a massive activation of XO is a major source of ROS in endothelial cells, and COX may contribute to this oxidative stress. Cytochrome P450 accounts for the production of ROS in lung epithelial cells, and NADPH oxidase accounts for ROS production in neutrophils that have infiltrated the affected tissues. The production of potent RNS appears to be more ubiquitous because a number of inflammatory mediators may lead to the induction of NOS in several cell types. In addition, the antioxidant mechanisms of the cell may be compromised and may further favor the accumulation of reactive species.
an important mechanism of reperfusion injury. These reactive species include highly unstable oxygen and nitrogen molecules, such as superoxide (O2–), hydrogen peroxide (H2O2), oxidized lipoproteins, lipid peroxides, nitric oxide (NO), and peroxynitrite (ONOO) (3). ROS and RNS can be formed by several mechanisms; they can be produced by a reduction of molecular oxygen in altered mitochondria; by enzymes such as xanthine oxidase (XO), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (also called Nox), cytochrome P450, nitric oxide synthase (NOS), and cyclooxygenase (COX); and by auto-oxidation of catecholamines. The source of reactive species varies from cell to cell and from stage to stage during the ischemia and reperfusion event. For example, in myocytes, but to a much lesser extent than in other cell types, the production of oxyradicals starts early in the ischemic period in altered mitochondria and is further enhanced during reperfusion. At reperfusion, a massive activation of XO is a major source of ROS in endothelial cells, and COX may contribute to this oxidative stress. Cytochrome P450 accounts for the production of ROS in lung epithelial cells, and NADPH oxidase accounts for ROS production in neutrophils that have infiltrated the affected tissues. The production of potent RNS appears to be more ubiquitous because a number of inflammatory mediators may lead to the induction of NOS in several cell types. In addition, the antioxidant mechanisms of the cell may be compromised and may further favor the accumulation of reactive species.These reactive species mediate tissue injury by two main mechanisms: (a) directly, by inducing the damage of important cellular macromolecules, and (b) indirectly, by activating signal transduction pathways that regulate the inflammatory and innate immune responses, and programmed cell death by apoptosis.
Sources of Reactive Oxygen and Nitrogen Species
Mitochondria
The generation of ROS starts during ischemia in the mitochondria. Although it might be expected that ischemia, leading to a low intracellular partial pressure of oxygen, would decrease oxyradical production, the paradoxic increase in ROS is secondary to the altered redox balance of the mitochondria. With ischemia, the metabolic reduction of the adenine nucleotide pool leaves the respiratory chain cytochromes in a more fully reduced state, allowing them to directly transfer (i.e., “leak”) electrons to the residual molecular oxygen entrapped within the inner mitochondrial membrane. This electron leakage appears capable of producing large amounts of superoxide radical. While reintroduction of oxygen with reperfusion reenergizes the mitochondria, the low ADP content also allows further electron leakage, thus enabling more reactions with molecular oxygen and yielding the excessive “burst” of ROS.
Within a few minutes of reperfusion, the pHi returns to normal values. However, in this altered milieu of calcium overload, high phosphate concentrations, and depletion of adenine nucleotides, the pH normalization favors the opening of the mitochondrial permeability transition pore, a highconductance channel whose opening leads to an increase of mitochondrial inner membrane permeability to solutes with molecular masses up to 1500 Da. This event plays a major role in the progression toward cell death. The opening of these pores releases stored calcium into the cytosol, and dissipation of the mitochondrial inner transmembrane potential uncouples the electron transport system from ATP hydrolysis. These events further abolish mitochondrial ATP production, leading to energy failure, enhanced production of ROS, and the release of apoptotic factors, thus contributing to cell death by apoptosis (4,5).
Xanthine Oxidoreductase System and NADPH Oxidase Systems
The xanthine oxidoreductase (XOR) enzyme system is an important enzymatic complex for the catabolism of purines, catalyzing the oxidation of hypoxanthine to xanthine and the oxidation of xanthine to uric acid. The system consists of two interconvertible forms, XO and xanthine dehydrogenase (XDH) (6). During ischemia, XDH is converted to the oxidase form by a protease activated by the intracellular overload of calcium. At the same time, degradation of ATP leads to an accumulation of hypoxanthine in the ischemic tissue. During reperfusion, with the presence of large quantities of molecular oxygen and high concentrations of hypoxanthine, XO yields a burst of superoxide (Fig. 21.4). During ischemia, the intracellular acidic conditions also allow the XOR enzyme system to catalyze the reduction of nitrates and nitrites to nitrites and NO, respectively, thus contributing to nitrosative stress.
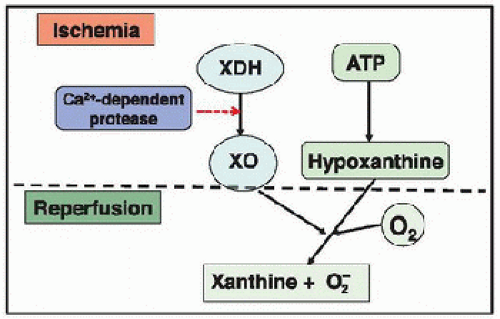 FIGURE 21.4. Superoxide production by xanthine oxidase. During ischemia, xanthine dehydrogenase (XDH) is converted to xanthine oxidase (XO) by a protease activated by the intracellular overload of calcium. At the same time, degradation of ATP leads to accumulation of hypoxanthine in the ischemic tissue. At reperfusion, large quantities of molecular oxygen are reintroduced into the cell. Both molecular oxygen and hypoxanthine then serve as substrates for XO to produce superoxide. |
Furthermore, ischemia and reperfusion in any tissue leads to the release of XOR into the circulation. Elevated plasma levels of XOR have been reported in adults subjected to limb ischemia-reperfusion (7) or liver transplantation (8). In critically ill infants and children with shock subsequent to severe sepsis, trauma, or major surgery, high XO activity has been detected in urine. This elevation in the XO activity index appears to correlate with high blood levels of oxidized glutathione (a marker of oxidative stress) and with the clinical symptoms of an excessive proinflammatory response (9). It has been suggested that, once in the circulation, XOR may contribute to the development of MODS. These circulating levels may, in fact, bind to the endothelial cells of distant sites and contribute to the initiation of oxidative damage in organs remote from the original ischemic tissue (6). The hypothesis that generation of superoxide by XO may play an important pathogenetic role in reperfusion injury has been supported by experimental studies in animals demonstrating that the inactivation of XO with allopurinol ameliorates reperfusion-induced tissue damage (10,11). A recent clinical study reported that allopurinol treatment might afford neuroprotective effects in moderately asphyxiated infants (12). Despite this human study and the beneficial effects reported in animal models, there is no other evidence of favorable effects of XOR inhibition in human conditions of reperfusion injury (10,11,13).
The NADPH oxidase family consists of seven Nox proteins: Nox1 to Nox5, Duox1 and Duox2. These proteins differ in their cell type expression, subunit composition, and function. In addition to phagocytic cells and neutrophils, where they mediate bacterial killing by producing a burst of superoxide anion, Nox enzymes are highly expressed in endothelial cells, vascular smooth muscle cells, cardiomyocytes, and renal tubular cells (14). A considerable number of experimental studies on mice genetically deficient of Nox proteins or their complex subunits suggest that this class of enzymes contribute to ischemia and reperfusion injury. Nox2 appears to contribute to lung, and brain reperfusion injury, as well as the remodeling process in the heart after infarction (14). Contribution of Nox enzymes in ischemia and reperfusion injury is suggested by clinical data demonstrating an upregulation of Nox2 and Nox5 in cardiomyocytes and intramyocardial blood vessels of patients who had died after myocardial infarction (15,16). Other clinical studies have reported that platelet-derived microparticles or exosomes, which were obtained from patients with septic shock, contained several Nox subunits and appeared to contribute to vascular cell apoptosis (17). At the present, there are no specific Nox inhibitors available. It has been reported that statins block activation of the Nox2 in addition to their HMG-CoA reductase inhibitory action (18,19); and polyphenols and anthocyanins, apart from their direct scavenging properties, can prevent Nox expression and reduce activity (20,21). However, whether the Nox system may be a therapeutic target in shock and reperfusion injury needs to be investigated in preclinical and clinical studies.
NOS Pathway
NO is a highly reactive gas that is synthesized from L-arginine by a family of enzymes referred as to NOSs. Three isoforms of NOS have been characterized: neuronal (nNOS, or type 1), inducible (iNOS, or type 2), and endothelial (eNOS, or type 3) (22). The eNOS and nNOS isoforms are constitutively expressed, are calcium/calmodulin dependent, and produce low amounts of NO. The designation of these constitutive isoforms does not strictly reflect their tissue expression because eNOS and nNOS are not only found in endothelial and neuronal cells but also in other cell types, including cardiomyocytes and muscle cells. The iNOS isoform is a calcium/calmodulin-independent enzyme that is responsible for the production of large amounts of NO. Although not detected under normal conditions, the expression of the iNOS isoform is induced in response to microbial products, inflammatory cytokines, and growth factors in almost every cell type during diverse pathologic conditions, including sepsis, hemorrhagic shock, trauma, and ischemia (22).
Under normal conditions, the constitutive forms of NOS release low concentrations of NO, which are critical to normal physiology. For example, the nNOS-derived NO acts as a neurotransmitter and a second messenger. The eNOS-derived NO is the physiologic mediator of normal vascular tone. Once formed by vascular endothelial cells, NO diffuses to adjacent smooth muscle cells, activates soluble guanylate cyclase, produces cyclic guanosine monophosphate (cGMP), and reduces intracellular calcium concentration, thus resulting in vasodilatation. Apart from its direct vasodilatory effect, the endothelium-derived NO also maintains normal tissue perfusion and vascular permeability by inhibiting platelet aggregation, leukocyte adherence, and smooth muscle proliferation in the vascular system. In cardiomyocytes, under physiologic conditions, both constitutive forms of NOS (types 1 and 3) have been described to release NO, which regulates cardiac function through their direct effects on several aspects of cardiomyocyte contractility, from the fine regulation of excitation -contraction coupling (with positive inotropic and lusitropic effects) to modulation of autonomic signaling and mitochondrial respiration (22,23).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


