Shock
Emanuel P. Rivers, David Amponsah, John Gallien, and Anja K. Jaehne
More than 1.5 million cases of shock present to United States (US) emergency departments (EDs) each year, and the number is increasing, particularly in patients older than 65 years. The incidence of septic shock, for example, has increased markedly for the past 10 years, with an annualized increase of 8.7% and more than 600,000 cases presenting to EDs each year in US (1). The annual economic burden for the care of patients in US with septic shock alone is more than $60 billion. Mortality from shock remains high despite better understanding of the underlying pathophysiology and aggressive treatment. Approximately 30% to 45% of patients in septic shock and 60% to 90% of those with cardiogenic shock die within 1 month of presentation. Over the last decade, the illness severity and duration for shock patients has significantly increased in EDs in US (2,3). Because of this, ED management is a central role in improving outcomes for these fragile patients.
Shock is defined as an impairment of circulatory function leading to global tissue hypoxia. The presentation may be as subtle as mild decompensated heart failure (4,5) or as obvious as the ultimate shock state of cardiac arrest. It is estimated that 12% of patients with undetected severe sepsis and septic shock are admitted to general medical floors, which is frequently associated with morbid and lethal consequences after hospital admission (6,7). Irrespective of the type of ED presentation, timely diagnosis and aggressive intervention prevents complications and significantly improves survival (7), especially in the ED setting (7).
PHYSIOLOGY
The principles of oxygen delivery (DO2) and utilization are paramount to the understanding of shock. A maximum of four molecules of oxygen are loaded onto each molecule of hemoglobin as it passes through the lungs. If all available oxygen sites are occupied (four per molecule of hemoglobin), then the arterial oxygen saturation (SaO2) is 100% (Fig. 4.1). Arterial oxygen concentration (CaO2) is the amount of oxygen bound to hemoglobin plus the amount dissolved in plasma. Oxygen delivery (DO2) is the product of the CaO2 and cardiac output (CO). Oxygen consumption (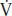 O2) reflects a sensitive balance between supply and demand. Normally, the tissues consume about 25% of the oxygen carried on hemoglobin, and venous blood returning to the right heart is about 65% to 75% saturated venous oxygen saturation (SVO2). When oxygen supply is insufficient to meet demand, the first compensatory mechanism is an increase in CO. If the increase in CO is inadequate, the amount of oxygen extracted from hemoglobin by the tissues increases (systemic oxygen extraction or OER), reflected in a decrease in SVO2.
O2) reflects a sensitive balance between supply and demand. Normally, the tissues consume about 25% of the oxygen carried on hemoglobin, and venous blood returning to the right heart is about 65% to 75% saturated venous oxygen saturation (SVO2). When oxygen supply is insufficient to meet demand, the first compensatory mechanism is an increase in CO. If the increase in CO is inadequate, the amount of oxygen extracted from hemoglobin by the tissues increases (systemic oxygen extraction or OER), reflected in a decrease in SVO2.
FIGURE 4.1 Oxygen saturation, content, and delivery.
When compensatory mechanisms (increased OER or decrease in SvO2 less than 50%) fail to correct the imbalance between tissue supply and demand, anaerobic metabolism occurs, resulting in the formation of lactic acid (normally between 0.5 and 1.9 mmol/L). Most cases of lactic acidosis are caused by inadequate DO2, but lactic acidosis occasionally can develop from an excessively high oxygen demand, as in status epilepticus or other nonhypoxic causes of lactic acidosis. In other cases, lactic acidosis occurs because of impairment in tissue oxygen utilization, as in septic shock, cyanide poisoning, and immediately after resuscitation from cardiac arrest. A normal SVO2 and adequate DO2 with an elevated lactate indicate such an impairment. An elevated lactate is a marker of impaired DO2 or utilization and correlates with short-term prognosis of critically ill patients in the ED (8,9). However, a normal lactate is not infrequent in patients with shock (10,11). SVO2 can also be used as a measure of the balance between tissue oxygen supply and demand. SVO2 is measured through a pulmonary artery catheter, but similar information can be obtained from the central venous oxygen saturation (SCVO2), which correlates well with SVO2 and is more easily obtained in the ED setting (12).
Thus, shock is circulatory insufficiency caused by tissue DO2 that is insufficient to meet oxygen demands. The result of shock is global hypoperfusion or tissue hypoxia and is manifested by a decreased venous oxygen content and metabolic acidosis (lactic acidosis). Global tissue hypoxia alone can independently activate the inflammatory response and serve as a comorbid variable in the pathogenesis of all forms of shock (13). The failure to diagnose and treat global tissue hypoxia in a timely manner leads to an accumulation of an oxygen debt, inflammation (14), organ dysfunction, and increased mortality.
CLINICAL PRESENTATION
The clinical presentation of shock can range from dramatic to subtle. Shock can be instantly apparent, as with acute myocardial infarction, anaphylaxis, or hemorrhage, whereas some patients may have few symptoms other than generalized weakness, lethargy, or altered mental status. Symptoms that suggest volume depletion include bleeding, vomiting, diarrhea, excessive urination, insensible losses as a result of fever, or orthostatic light-headedness. A history of cardiovascular disease is important, particularly episodes of chest pain or symptoms of congestive heart failure (particularly systolic dysfunction). Prior neurologic diseases (e.g., Parkinson) can render patients more susceptible to complications from hypovolemia because of autonomic dysfunction. Use of medications, both prescribed and nonprescribed, is important. Some medications cause volume depletion (e.g., diuretics), whereas others depress myocardial contractility (e.g., β-blockers and calcium channel blockers) and others are vasodilators (e.g., niacin, erectile dysfunction, or prostatic hypertrophy drugs). The previous use of corticosteroids may predispose the patient to adrenal insufficiency. The possibility of an anaphylactic reaction to a new medication should also be considered.
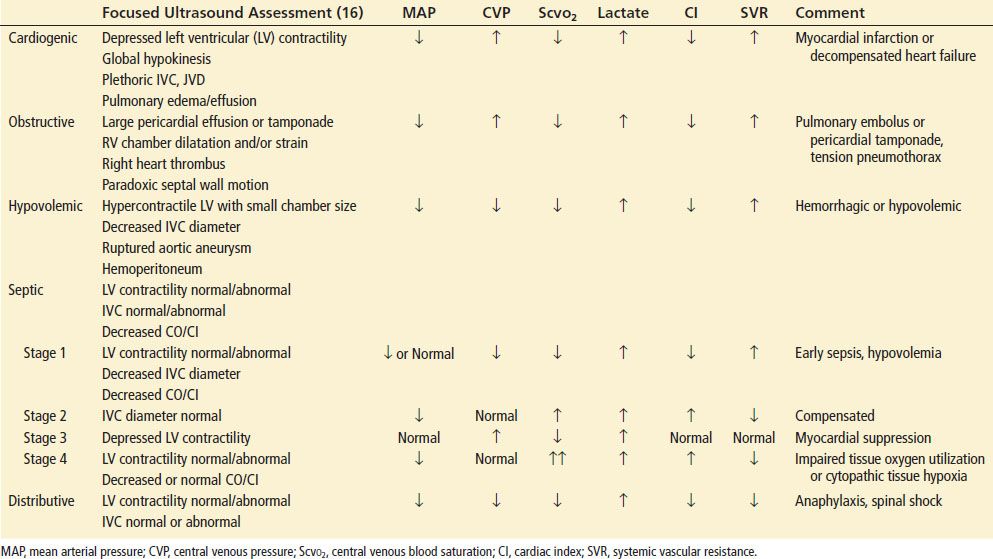
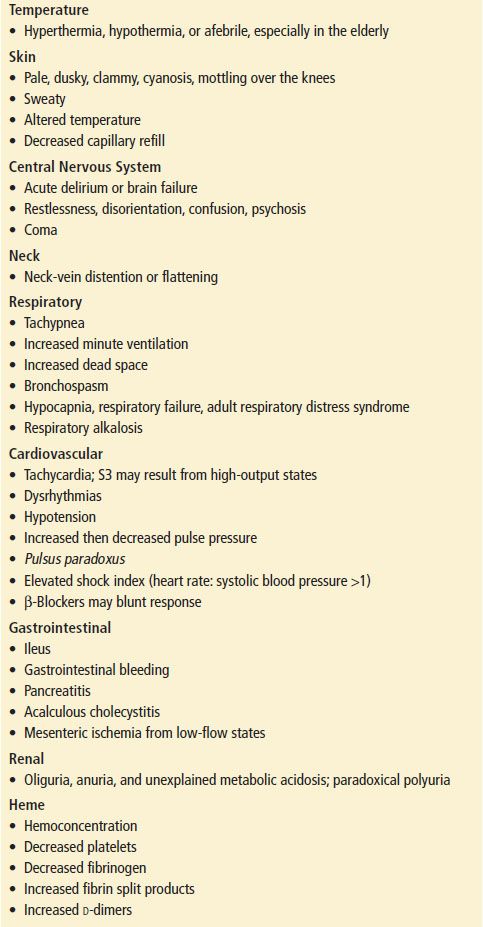
The clinical presentation of shock varies depending on the precipitating cause such as cardiogenic, hypovolemic, obstructive, or distributive. Although not specific, physical findings taken as a composite may be useful in the assessment of patients in shock (Table 4.1). No single vital sign or physical finding is diagnostic of shock, as vital signs are frequently insensitive in detecting and assessing the severity of tissue hypoperfusion. Measurement of blood pressure can be particularly difficult because of peripheral vascular disease, tachycardia with a small pulse pressure, arteriovenous dialysis fistulas and dysrhythmias such as atrial fibrillation. Shock is usually, but not always, associated with systemic arterial hypotension, which is systolic blood pressure <90 mm Hg. Nonsustained hypotension, or a single episode, carries a significantly increased risk of death during hospitalization, which makes it a clinically significant and meaningful event (15).
TABLE 4.1
Clinical Findings in Shock
Because blood pressure is the product of flow and resistance (mean arterial pressure [MAP] = CO × systemic vascular resistance [SVR]), the blood pressure may not fall in the presence of decreased CO if there is an increase in peripheral vascular resistance (Fig. 4.2). The insensitivity of blood pressure in detecting inadequate flow to tissues and global tissue hypoxia has been repeatedly confirmed. Thus, shock may occur with a normal blood pressure, and hypotension may occur without shock.
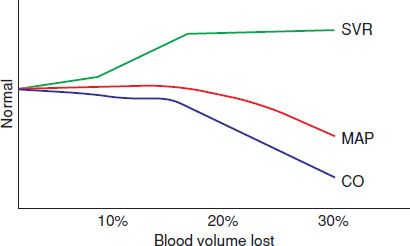
FIGURE 4.2 Relationship of SVR (systemic vascular resistance), MAP (mean arterial pressure), and CO (cardiac output) with volume loss.
The onset of shock provokes a myriad of autonomic responses, many of which serve to maintain perfusion pressure to vital organs. Stimulation of the carotid baroreceptor stretch reflex activates the sympathetic nervous system leading to (a) arteriolar vasoconstriction, resulting in redistribution of blood volume from the skin, skeletal muscle, kidneys, and splanchnic viscera; (b) an increase in heart rate and contractility that increases CO; (c) constriction of venous capacitance vessels, which augments venous return; (d) release of the vasoactive hormones epinephrine, norepinephrine, dopamine, and cortisol to maintain arteriolar and venous tone; and (e) release of antidiuretic hormone and activation of the renin–angiotensin axis to enhance water and sodium conservation to maintain intravascular volume. These compensatory mechanisms attempt to maintain DO2 to the most critical organs: the coronary and cerebral circulation. During this process, blood flow to other organs, such as to the kidneys and gastrointestinal tract, may be compromised, leading to organ failure.
The cellular response to decreased DO2 is adenosine triphosphate (ATP) depletion, leading to ion pump dysfunction, influx of sodium, efflux of potassium, and reduction in membrane resting potential. Cellular edema occurs secondary to increased intracellular sodium while cellular membrane receptors become poorly responsive to the stress hormones insulin, glucagon, cortisol, and catecholamines. As shock progresses, lysosomal enzymes are released into the cells with subsequent hydrolysis of membranes, deoxyribonucleic acid, ribonucleic acid, and phosphate esters. As the cascade of shock continues, the loss of cellular integrity and the breakdown in cellular homeostasis result in cellular death (Fig. 4.3). The failure of the cell to utilize oxygen or perform cellular respiration is impaired, and thus endogenous heat generation is impaired. This process gives rise to hypothermia, a common clinical feature of shock. These pathologic events give rise to the metabolic features of hemoconcentration, hyperkalemia, hyponatremia, prerenal azotemia, metabolic acidosis, hyper- or hypoglycemia, and lactic acidosis.
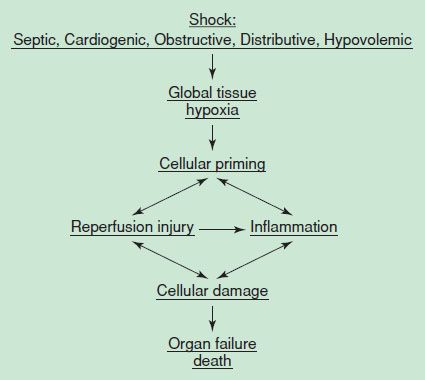
FIGURE 4.3 Mechanism of cell death.
The clinical presentation of early shock is often characterized as a syndrome called the systemic inflammatory response syndrome (SIRS). SIRS is defined as the presence of two or more of the following features: (a) temperature >38°C (100.4°F) or <36°C (96.8°F); (b) heart rate >90 beats/min; (c) respiratory rate >20 breaths/min; and (d) white blood cell count >12 × 109/L, <4 × 109/L, or with >10% immature forms or bands. SIRS may occur in any shock state. The stimulus for SIRS is a complex elaboration of humoral and cellular pro- and anti-inflammatory products, which induces a panendothelial cell disruption or generalized inflammation within the vast capillary network in each organ. The hemodynamic consequence of SIRS is the generation of global tissue hypoxia from hypovolemia, vasodilation, and myocardial suppression. There is an increase in oxygen demands secondary to fever or an increased work of breathing. Cytopathic tissue hypoxia develops when there is a defect in the peripheral utilization of oxygen in the tissues.
As SIRS progresses, organ dysfunction and the multiorgan dysfunction syndrome (MODS) ensue. This syndrome is characterized by organ failures such as acute lung injury, adult respiratory distress syndrome, disseminated intravascular coagulation, hepatic failure, adrenal and renal failure, among others. Early detection and interruption of these hemodynamic perturbations are key for improving survival. Table 4.2 compares shock states.
TABLE 4.2
Comparison of Shock States