INTRODUCTION AND EPIDEMIOLOGY
Sepsis is a heterogeneous syndrome characterized by widespread inflammation and potential organ harm initiated by a microorganism. Although gram-positive and gram-negative bacteria account for the majority of cases, fungi, viruses, mycobacteria, rickettsiae, and protozoans can trigger sepsis. Invasion of the blood is not necessary to develop or identify sepsis, which is determined by the host response to the infectious insult. As sepsis severity increases, a multifactorial series of events lead to impairments in oxygen delivery, secondary to macro- and microvascular malperfusion, as well as direct cellular damage secondary to inflammation. Eventually, multisystem organ failure occurs, and mortality is high.
The varying clinical presentation, differences in coding, and methodologic differences between studies lead to a wide range of estimates of the annual incidence of severe sepsis, ranging from 300 to 1000 cases/100,000 persons per year.1 Over 500,000 patients each year present to the ED with suspected severe sepsis, the largest group of all sepsis patients hospitalized. The incidence is increasing and is multifactorial in origin; a key component of this increase relates to an aging patient population, which is not surprising given the fact that sepsis incidence increases >100-fold with age (0.2 per 1000 in children age 10 to 14 years to 26.2 per 1000 in those >85 years of age).2
The mean ED length of stay of a patient with sepsis is 5 hours. Once admitted, more than half of patients with severe sepsis will require care in an intermediate or intensive care unit,2 where it represents the leading cause of death. Despite advances in care, mortality rates from severe sepsis remain high, with approximately 20% dying3 during hospitalization in optimal clinical trial scenarios4,5; this rate approaches 50% when considering the sicker subset of those with septic shock.6 These mortality rates exceed many other high-visibility acute care conditions such as acute myocardial infarction,7 massive pulmonary embolism,8 and cerebrovascular accident.9 Morbidity is high and prolonged, and a long-term deficit in cognition and functioning is common.10 Finally, sepsis care is costly; estimates from 10 years ago suggest a mean case cost of $22,100 with annual national costs reaching $16.7 billion,2 figures that have certainly increased in the last decade.
Since 1987, gram-positive bacteria outside of the surgical setting are the predominant pathogens of sepsis.11 With the rise of antimicrobial resistance, methicillin-resistant Staphylococcus aureus, vancomycin-resistant Enterococcus, and other multidrug-resistant organisms are more common.12 Similarly, the incidence of fungi as the source has risen, particularly in immunosuppressed patients. The most likely causative microorganism varies based on the likelihood of exposure to drug-resistant microorganisms (due to recent healthcare exposures) and the anatomic site of infection, with pneumonia, intra-abdominal, urinary, and skin/soft tissue being the most common locations.
DEFINITIONS
Sepsis syndromes are a continuum, with specific definitions regularly updated to include children13,14 (Table 151-1). Although the definitions provide a conceptual and practical framework for recognition of the systemic inflammatory response to infection, definitions often are not sensitive or specific in the real-world clinical setting. In general, sepsis is defined as suspected or confirmed infection with evidence of systemic inflammation (demonstrated either through evidence of the systemic immune response syndrome or laboratory abnormalities), whereas severe sepsis is generally defined as sepsis plus evidence of new organ dysfunction thought to be secondary to tissue hypoperfusion. Septic shock exists when cardiovascular failure occurs, evidenced as persistent hypotension or need for vasopressors despite adequate fluid resuscitation; this latter category has a particularly poor prognosis.
Systemic Inflammatory Response Syndrome (SIRS) criteria: 1. Fever (temperature >38.3°C) or hypothermia (temperature <36°C) 2. Pulse rate (>90 beats/min or >2 SDs above the normal value for age) 3. Tachypnea (respiratory rate >20 breaths/min) 4. Leukocytosis (WBC >12,000 cells/μL) or leukopenia (WBC <4000 cells/μL), or normal WBC with >10% immature forms Sepsis:Infection (documented or suspected), and some of the following: General Parameters: Fever (temperature >38.3°C) Hypothermia (temperature <36°C) Pulse rate (>90 beats/min or >2 SDs above the normal value for age) Tachypnea Altered mental status Significant edema or positive fluid balance (>20 mL/kg during 24 h) Hyperglycemia (plasma glucose >140 milligrams/dL or 7.7 mmol/L) in the absence of diabetes Inflammatory Parameters: Leukocytosis (WBC >12,000 cells/μL) Leukopenia (WBC <4000 cells/μL) Normal WBC with >10% immature forms Plasma C-reactive protein (CRP) >2 SDs above the normal value Plasma procalcitonin >2 SDs above the normal value Hemodynamic Parameters: Arterial hypotension (SBP <90 mm Hg, MAP <70 mm Hg, or an SBP decrease >40 mm Hg in adults or <2 SDs below normal for age) Organ Dysfunction Parameters: Arterial hypoxemia (PaO2/FIO2<300) Acute oliguria (urine output <0.5 mL/kg per hour for at least 2 h despite adequate fluid resuscitation) Creatinine level increase >0.5 milligrams/dL Coagulation abnormalities (INR >1.5 or aPTT >60 s) Ileus (absent bowel sounds) Thrombocytopenia (platelet count <100,000 cells/μL) Hyperbilirubinemia (plasma total bilirubin >4 milligrams/dL) Tissue Perfusion Parameters: Hyperlactatemia (above upper limits of laboratory normal levels) Decreased capillary refill or mottling Severe Sepsis:Sepsis-induced tissue hypoperfusion or organ dysfunction (any of the following thought to be due to infection) Sepsis-induced hypotension Lactate level above upper limits of laboratory normal levels Urine output <0.5 mL/kg per hour for at least 2 h despite adequate fluid resuscitation Acute lung injury with PaO2/FIO2<250 in the absence of pneumonia as infectious source Acute lung injury with PaO2/FIO2<200 in the absence of pneumonia as infectious source Creatinine level >2.0 milligrams/dL Bilirubin level >2 milligrams/dL Platelet count <100,000 cells/μL Coagulopathy (INR >1.5) |
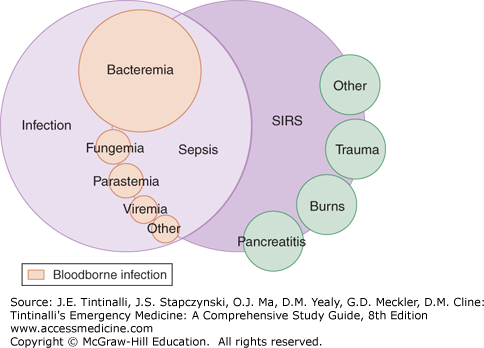
Having systemic inflammatory response syndrome criteria does not confirm the presence of infection or sepsis because these features are shared by many other noninfectious conditions such as trauma, pancreatitis, and burns (Figure 151-1). The systemic inflammatory response syndrome response is not a diagnosis or a good indicator of outcome; it is a crude means of stratification of patients with systemic inflammation. In a prospective study of the epidemiology of patients demonstrating systemic inflammatory response syndrome (infectious and noninfectious), mortality rates were 3% in patients without systemic inflammatory response syndrome, 6% in those meeting two criteria, 10% in those meeting three criteria, and 17% in those meeting all four criteria.14 Death rates were similar whether or not patients had positive blood cultures.
Complementary methods of classification of sepsis based primarily on physiologic abnormalities include the third Acute Physiology and Chronic Health Evaluation acuity system,15 the Sequential Organ Failure Assessment score, and the Mortality in Emergency Department Score16; each allows for prognostication. These scoring systems are typically limited to research and are not for guiding patient care. Any evidence of septic shock confers the highest mortality risk assessment.
Serum lactate across a wide spectrum17 provides excellent prognostic data in those with sepsis, but is not a singular test to diagnose or exclude sepsis. Lactate has traditionally been attributed to anaerobic metabolism secondary to tissue hypoperfusion; it also accumulates from changes in aerobic metabolism that occur in response to widespread inflammation.18 As lactate increases, the risk of mortality rises, with the degree of lactate elevation and hypotension being independent predictors of mortality.19,20 Specifically, 28-day mortality rate associated with modest elevation of serum lactate to 2 to 4 mmol/L approaches 15% even in those patients without hypotension,21 and the rate continues to rise with increasing lactate levels. In general, venous or arterial lactate levels on either point-of-care bedside devices or measured in a central laboratory are useful if done with good clinical technique22; even better are serial levels using the same method of sampling (arterial or venous) to identify improvement that mirrors clinical responses.22 This is called lactate clearance and is defined as a drop over time; lactate clearance provides an independent prognostic value in addition to vital signs and other measurements. Patients with sepsis who do not clear lactate (adequate is at least a 10% drop with therapy over the first few hours) have a higher mortality rate than patients who do clear the lactate.23,24 The major limitation is that lactate is nonspecific and can be elevated in a number of conditions, and it must be used together with clinical judgment.
PATHOPHYSIOLOGY
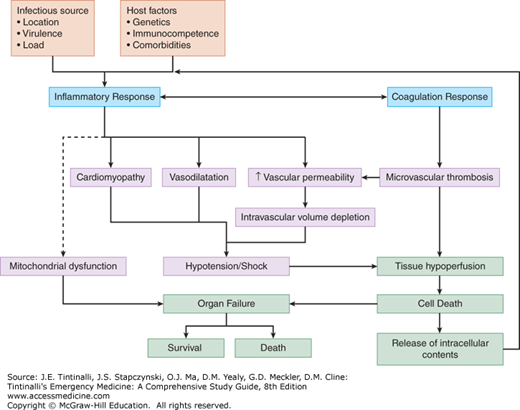
In sepsis, the host immune response fails to control and/or overreacts to invasive pathogens, leading to two critical events.25,26 The first event involves marked abnormalities in the inflammatory response in the host. The host response typically varies from a hyperinflammatory response in the early stages of sepsis, to a blunted inflammatory response in the later stages of sepsis, which leads to an increased risk of secondary hospital-acquired infections. The blunted inflammatory response results in programmed death of key immune, epithelial, and endothelial cells, leading to tissue injury and perpetuating multiorgan dysfunction.
The second event is an imbalance in procoagulant and anticoagulant functioning; in the most extreme of situations, this results in the clinical syndrome of disseminated intravascular coagulation. Disseminated intravascular coagulation results in micro- and macrovascular clot formation, impaired microvascular tissue perfusion, and thrombosis of small vessels. Notably, subclinical disseminated intravascular coagulation is often lurking despite relatively normal basic laboratory findings, with impaired microvascular perfusion (detecTable in research settings) that is associated with a worse prognosis.27 Continued microvascular ischemia likely contributes to organ failure, as well as to the release of proinflammatory intracellular contents, which further stimulates the innate immune response and perpetuates the underlying pathology.28 As these events progress, they intensify the inflammatory response and a destructive cycle ensues, as illustrated in Figure 151-2. At this time, there are no specific therapies to remedy microvascular dysfunction.
CLINICAL FEATURES
While some presentations of severe sepsis are immediately clinically apparent, sepsis can present in a subtle or occult manner, particularly early in the course. Given that systemic inflammation produces physiologic changes, vital sign abnormalities—notably fever, hypotension, and/or tachycardia—are a hint to sepsis, recognizing that many patients with these findings may have another cause. In ED patients with undifferentiated hypotension, 40% will ultimately have an infectious cause of symptoms.29
Although traditionally sepsis is categorized as an example of distributive shock (peripheral vasodilation evidenced by warm extremities with a compensatory increased cardiac output), this presentation does not accurately describe all patients with sepsis. ED patients with sepsis are often volume depleted from decreased intake and increased fluid losses (from emesis, diarrhea, or insensible losses associated with fever and tachypnea). Intravascular volume depletion directly affects preload, cardiac output, and ultimately peripheral perfusion. Further complicating matters is septic cardiomyopathy, a reversible process characterized by impaired systolic function and diastolic relaxation.30 Finally, the combination of intravascular volume depletion and septic cardiomyopathy may manifest as “cold shock,” impaired peripheral perfusion and cool extremities.
Widespread inflammation secondary to sepsis commonly affects pulmonary function even in the absence of pneumonia. Acute lung injury is common and may result in acute respiratory distress syndrome, which is characterized by new lung edema from increased alveolar and capillary permeability. Classification of acute respiratory distress syndrome is based primarily on the degree of hypoxemia.31 The three mutually exclusive categories are mild (PaO2 divided by fraction of inspired oxygen [FIO2] of 200 to 300), moderate (PaO2/FIO2 of 100 to 200), and severe (PaO2/FIO2 <100), within 1 week of a clinical insult or change in respiratory status coupled with new bilateral pulmonary infiltrates on chest radiographs not explained by effusions, lung collapse, or nodules and not fully explained by heart failure or volume overload. Clinically, severe refractory hypoxemia, noncompliant lungs noted on mechanical ventilation, and a chest radiograph showing bilateral pulmonary alveolar infiltrates suggest the diagnosis. Mortality increases from 27% to 45% with increasing severity of acute respiratory distress syndrome.
The kidney is another common sepsis target; acute kidney injury can present with azotemia, oliguria, or anuria. Factors increasing acute kidney injury risk include pre-existing kidney dysfunction or vasculopathy, depth and duration of hypotension, dehydration, and use of nephrotoxic substances (e.g., aminoglycoside antibiotics, nonionic intravenous contrast). Renal ischemic injury from hypoperfusion is a major factor in the pathogenesis of acute kidney injury in sepsis, although toxic products resulting from neutrophil-endothelial interactions, endothelial damage by various mediators, reperfusion injury, and microvascular thrombosis also contribute.
The most frequent hepatic abnormality is cholestatic jaundice, although it occurs infrequently. Increased concentrations of transaminase, alkaline phosphatase (one to three times the normal level), and bilirubin (usually not >10 milligrams/dL) may be observed. Marked elevations of transaminases or bilirubin are less common unless septic shock is present; if seen, consider a biliary source of infection. Smaller elevations in liver function tests can result from intermittent or prolonged macro- or microvascular hypoperfusion and ischemia or can be secondary to direct endotoxin, cytokines, or immune complex damage. Red blood cell hemolysis from microvascular coagulation can also rarely cause jaundice.
The most common GI manifestation of sepsis is ileus, which may persist for days after shock resolves. Major blood loss secondary to upper GI bleeding is rare in septic patients. Minor GI blood loss within 24 hours of developing severe sepsis can result from painless erosions in the mucosal layer of the stomach or duodenum.
Multiple abnormalities are possible in the hematologic system in the setting of sepsis, including neutropenia or neutrophilia, thrombocytopenia, or disseminated intravascular coagulation. Neutrophilic leukocytosis with a “left shift” results from demargination and release of newer granulocytes from the marrow storage pools. However, the presence of excessive bands (the immature neutrophil) is neither sensitive nor specific for infection. Neutropenia occurs rarely and is associated with an increase in mortality; it results from increased peripheral use of neutrophils, damage to neutrophils by bacterial by-products, or depression of marrow granulocyte production by inflammatory mediators. Functional neutropenia also creates a relative immunosuppression, particularly later in patients’ hospital course. Both red cell production and survival decrease during sepsis, but anemia is not expected unless it existed prior to the infection, the infection is extremely prolonged, or there is concomitant bleeding.
Thrombocytopenia may arise as a consequence of disseminated intravascular coagulation but is present in >30% of cases of sepsis even in the absence of overt disseminated intravascular coagulation, and lower platelet levels are associated with worse outcomes.32,33 Proposed mechanisms of thrombocytopenia include inhibition of thrombopoiesis, increased platelet turnover (due to a consumptive coagulopathy), increased endothelial adherence, and increased destruction secondary to immunologic mechanisms.
Finally, fulminant disseminated intravascular coagulation where clinical clotting and bleeding exist is rare but associated with a very poor prognosis.26 Occult disseminated intravascular coagulation, with microvascular flow abnormalities, subclinical increased activation of the coagulation system, and evidence of increased fibrinogen split products in the absence of overt bleeding and clotting, is much more common. The activation of the hemostatic (clotting) system is due primarily to the activation of the extrinsic pathway of clotting. The fibrinolytic system is also activated in sepsis and plays an important role in limiting fibrin deposition in the microcirculation. The release of tissue plasminogen activator activates the fibrinolytic system, at least initially in sepsis. As sepsis progresses, there is an increased release of plasminogen activator inhibitor 1, which blocks plasmin generation and thus contributes to fibrin deposition in the microcirculation and subsequent multiple-organ failure. Laboratory studies suggesting the presence of disseminated intravascular coagulation include thrombocytopenia, prolonged prothrombin and activated partial thromboplastin values, decreased fibrinogen and antithrombin III levels, and increased fibrin monomer, fibrin split products, and D-dimer levels.
Sepsis induces multiple metabolic changes. Abnormalities in lactate metabolism due to tissue hypoperfusion with resultant anaerobic metabolism as well as increased aerobic production of lactate have been discussed previously (see “Systemic Inflammatory Response Syndrome Prognosis”). Hyperglycemia is seen even in patients without a history of diabetes; in this latter group, it is associated with a worse prognosis, in contrast to less impact of glucose elevations in those with known diabetes.34 Hypoglycemia with glucose levels as low as 10 to 20 milligrams/dL is reported but uncommon, and may result due to depletion of hepatic glycogen and inhibition of gluconeogenesis and/or adrenal insufficiency. Adrenal insufficiency can occur,35 caused by hypoperfusion of the adrenal glands, adrenal or pituitary hemorrhage, cytokine dysfunction of the adrenals, drug-induced hypermetabolism, inhibition of steroidogenesis by chemotherapeutics (e.g., ketoconazole), and desensitization of glucocorticoid responsiveness at the cellular level.
There are five potential cutaneous manifestations of sepsis: direct bacterial involvement of the skin and underlying soft tissues (cellulitis, erysipelas, and fasciitis); lesions from hematogenous seeding of the skin or the underlying tissue (petechiae, pustules, cellulitis, ecthyma gangrenosum); lesions from hypotension and/or disseminated intravascular coagulation (acrocyanosis and necrosis of peripheral tissues); lesions from intravascular infections (microemboli and/or immune complex vasculitis); and lesions caused by toxins (toxic shock syndrome). Look for any necrotizing or surgical source of sepsis, and recall that the toxic shock syndromes present in an overlapping fashion (see chapter 150, “Toxic Shock Syndromes”).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


