GENERAL APPROACH TO RESPIRATORY DISTRESS
Dyspnea is a subjective feeling of difficult, labored, or uncomfortable breathing, which patients often describe as “shortness of breath,” “breathlessness,” or “not getting enough air.”1 Dyspnea is frequently associated with other respiratory symptoms or signs. Tachypnea is rapid breathing. Orthopnea is dyspnea in the recumbent position. It is most often the result of left ventricular failure, but can also be seen with diaphragmatic paralysis or chronic obstructive pulmonary disease. Paroxysmal nocturnal dyspnea is orthopnea that awakens the patient from sleep, prompting an upright posture in order to resolve breathlessness. Trepopnea is dyspnea associated with only one of several recumbent positions. Trepopnea can occur with unilateral diaphragmatic paralysis, with ball-valve airway obstruction, or after surgical pneumonectomy. Platypnea is the opposite of orthopnea: dyspnea in the upright position. Platypnea results from the loss of abdominal wall muscular tone and, in rare cases, from right-to-left intracardiac shunting, as occurs from a patent foramen ovale. Hyperpnea is essentially hyperventilation and is defined as minute ventilation in excess of metabolic demand. Respiratory distress is a term used by the physician, combining the patient’s subjective sensation of dyspnea with signs indicating difficulty breathing. Ventilatory or respiratory failure occurs when the lungs and ventilatory muscles cannot move enough air in and out of the alveoli to adequately oxygenate arterial blood and eliminate carbon dioxide.
Dyspnea is a complex sensation that arises from the interaction of multiple pathophysiologic mechanisms.1,2 Sensory information about respiratory activity generated by multiple afferent receptors is integrated within the CNS at both the subcortical and cortical levels. The current explanation for the sensation of dyspnea is when imbalance exists among the inspiratory drive, efferent activity to the respiratory muscles, and feedback from these afferent receptors.
Dyspnea is a feature of several disorders seen in the ED (Table 62-1). The presence or degree of dyspnea is difficult to measure, although categorical scales (e.g., the Borg or Fletcher scales) and visual analog scales can be used to gauge response to therapy.1,3 Assess for evidence of impending respiratory failure: marked tachypnea and tachycardia; stridor; use of the accessory respiratory muscles, including the sternocleidomastoid, sternoclavicular, and intercostals; inability to speak normally as a consequence of breathlessness; agitation or lethargy as a consequence of hypoxemia; depressed consciousness due to hypercapnia; and paradoxical abdominal wall movement when the abdominal wall retracts inward with inspiration, indicating diaphragmatic fatigue. In patients with these signs, give oxygen and be prepared for more advanced measures (discussed elsewhere). Lesser degrees of dyspnea allow for a more detailed medical history, physical examination, and indicated ancillary tests.
Most Common Causes
| Most Immediately Life-Threatening Causes
|
Ask about recent infectious and environmental exposures that may impair respiratory function. Carefully question patients who require daily medications for symptom control about compliance and possible drug interactions.
Dyspnea is a prominent symptom of heart failure,4 and differentiating heart failure from pulmonary causes of dyspnea is an important and frequently difficult task. Treatment and prognosis differ, and embarking down the wrong pathway of treatment can have adverse consequences.5 Several findings can assist in this differentiation, although few of them are definitive by themselves (Table 62-2).6,7,8
| Finding | LR+ (95% CI) | LR– (95% CI) |
|---|---|---|
| Clinical gestalt | 4.4 (1.8–10.0) | 0.45 (0.28–0.73) |
| History | ||
| Heart failure | 5.8 (4.1–8.0) | 0.45 (0.38–0.53) |
| Myocardial infarction | 3.1 (2.0–4.9) | 0.69 (0.58–0.82) |
| Coronary artery disease | 1.8 (1.1–2.8) | 0.68 (0.48–0.96) |
| Symptoms | ||
| Paroxysmal nocturnal dyspnea | 2.6 (1.5–4.5) | 0.70 (0.54–0.91) |
| Orthopnea | 2.2 (1.2–3.9) | 0.65 (0.45–0.92) |
| Edema | 2.1 (0.92–5.00) | 0.64 (0.39–1.10) |
| Dyspnea on exertion | 1.3 (1.2–1.4) | 0.48 (0.35–0.67) |
| Physical examination | ||
| S3 gallop | 11.0 (4.9–25.0) | 0.88 (0.83–0.94) |
| Jugular venous distention | 5.1 (3.2–7.9) | 0.66 (0.57–0.77) |
| Hepatojugular reflex | 6.4 (0.81–51.00) | 0.79 (0.62–1.00) |
| S4 | 1.6 (0.47–5.50) | 0.98 (0.93–1.00) |
| Wheezing | 0.52 (0.38–0.71) | 1.3 (1.1–1.7) |
| Chest x-ray | ||
| Pulmonary venous congestion | 12.0 (6.8–21.0) | 0.48 (0.28–0.83) |
| Interstitial edema | 12.0 (5.2–27.0) | 0.68 (0.54–0.85) |
| Alveolar edema | 6.0 (2.2–16.0) | 0.95 (0.93–0.94) |
| Cardiomegaly | 3.3 (2.4–4.7) | 0.33 (0.23–0.48) |
| ECG | ||
| Atrial fibrillation | 3.8 (1.7–8.8) | 0.79 (0.65–0.96) |
| Any abnormal finding | 2.2 (1.6–3.1) | 0.64 (0.47–0.88) |
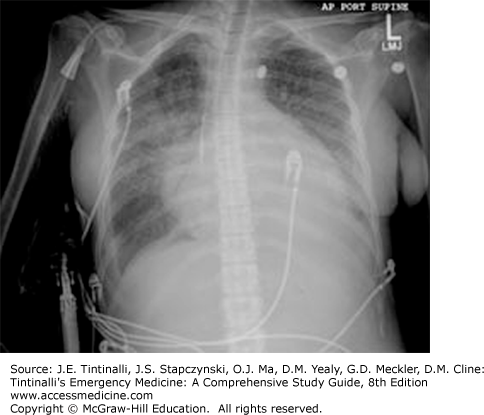
An S3 gallop on physical examination or pulmonary venous congestion/interstitial edema (especially with concomitant cardiomegaly) on chest x-ray strongly suggest heart failure as the cause of the dyspnea (Figure 62-1).6 The physician’s overall gestalt of the diagnosis, the presence of jugular venous distention on examination, and alveolar edema on chest x-ray suggest heart failure. Wheezing, dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea, and leg edema are not useful in discriminating between cardiac and pulmonary causes. Conversely, the absence of these findings does not exclude heart failure.
Pulse oximetry provides a rapid assessment of arterial oxygen saturation but is an insensitive screening test for disorders of gas exchange. Arterial blood gas analysis is more sensitive for detecting impaired gas exchange but cannot evaluate the work of breathing. Arterial blood gas testing can find the rare patient with dyspnea or tachypnea who exhibits no evidence of hypoxemia or pulmonary disease, suggesting hyperventilating from metabolic acidosis.
Bedside spirometric analysis (e.g., peak expiratory flow), especially if performed before and after bronchodilator therapy, can be used to diagnose dyspnea resulting from asthma or chronic obstructive pulmonary disease, but spirometry requires voluntary effort that might be difficult for dyspneic patients.
Negative inspiratory force can assess strength of the diaphragm and inspiratory muscles. Other potentially useful tests include an ECG and measuring the hemoglobin level. In most ED patients, the cause of dyspnea can be identified by the history, the physical examination, and these ancillary tests.
B-type natriuretic peptide (BNP) is a polypeptide secreted by ventricular myocytes in response to volume expansion and pressure overload; BNP elevates with any cause of overload, including heart failure, myocardial ischemia, pulmonary embolism, sepsis, chronic obstructive pulmonary disease, or any right heart strain. Serum levels of BNP or its precursor, N-terminal pro-BNP, are measured using two methods: an enzyme-linked immunosorbent assay and a radioimmunoassay; the enzyme-linked immunosorbent assay is more accurate than the radioimmunoassay.9
A normal BNP (<100 picograms/mL) or N-terminal pro-BNP (<500 picograms/mL) excludes heart failure in low and moderate pretest probability patients outside of “flash” settings.6,9,10 A high level (BNP >500 picograms/mL or N-terminal pro-BNP >2000 picograms/mL) is moderately useful for establishing the diagnosis of heart failure, although these elevations are rarely unsuspected after a careful history, exam, and chest radiograph.10,11 Overall, BNP measurement offers limited real help in assessing dyspneic patients,12 especially when values between 100 and 500 picograms/mL occur, which is common in the patient without a clear clinical syndrome.11,13,14
A chest radiograph may find a pulmonary abnormality, infiltrate, effusion, and pneumothorax.
Bedside lung US is an important tool in the assessment of acute dyspnea. It can differentiate acute decompensated heart failure from noncardiac causes of acute dyspnea with a sensitivity and specificity of about 97% and is superior to chest x-ray and natriuretic peptide determination.15,16 Bedside US can identify pleural effusion, pneumothorax, cardiac tamponade, cardiac functional abnormalities, pulmonary consolidation, and intravascular volume status (Figures 62-2,62-3,62-4).17,18
In severe dyspnea, the initial treatment goal is maintenance of the airway and oxygenation, seeking a partial pressure of alveolar oxygen (PaO2) > 60 mm Hg and/or arterial oxygen saturation (SaO2) ≥90%. Next, or in those with lesser dyspnea, treat the underlying disorder. Rarely are opioids or benzodiazepines used as dyspnea therapy, except in near terminal states for patient comfort.1,19
HYPOXIA AND HYPOXEMIA
Hypoxia is insufficient delivery of oxygen to the tissues. The amount of oxygen available to the tissues is a function of the arterial oxygen content (CaO2) comprising a small portion dissolved in plasma and a larger portion bound to Hb:
Oxygen delivery (DO2) is a product of arterial oxygen content and cardiac output (CO):
Tissue hypoxia occurs in states of low cardiac output, low Hb concentration, or low SaO2. The level of oxygen saturation in arterial Hb is, in turn, dependent on the PaO2, as determined by the oxygen-Hb dissociation curve. Hypoxemia is an abnormally low arterial oxygen tension (defined as a PaO2 <60 mm Hg). While hypoxemia is the most common cause of hypoxia, hypoxia and hypoxemia are not interchangeable; one can occur without the other. For example, in states of low PaO2 (hypoxemia) with concomitant polycythemia, the patient may have no tissue hypoxia. Alternatively, severely anemic patients may suffer tissue hypoxia despite a normal PaO2.
Relative hypoxemia is the term used when the arterial oxygen tension is lower than expected for a given level of inhaled oxygen. The degree of relative hypoxemia can be assessed by calculating the alveolar arterial (A-a) oxygen partial pressure gradient, [P(A-a)O2], which measures efficiency of oxygen transfer from the lungs to the circulation. Alveolar oxygen partial pressure is determined by the inhaled oxygen concentration (21% for room air), atmospheric pressure (760 mm Hg at sea level), and displacement by water vapor (47 mm Hg for full saturation) and carbon dioxide (CO2). Gas in the alveolus is fully saturated with water vapor, and the amount of alveolar oxygen is further reduced by CO2 that freely diffuses from the pulmonary capillaries in an amount determined by the ratio between oxygen consumption and CO2 production, termed the respiratory quotient (R), which is affected by diet. On a typical mixed diet, the R is 0.8. Alveolar oxygen breathing room air at sea level has a:
The A-a gradient at sea level for room air is a:
A simplified formula often used is:
A normal P(A-a)O2 is <10 mm Hg in young, healthy patients and increases with age, predicted by the formula:
This normal A-a gradient is for healthy, asymptomatic individuals measured in an upright or sitting position. The supine position alone, as well as many chronic cardiac or pulmonary diseases, may raise the A-a gradient. The supine position is a common ED patient position, impairing the assessment.
Hypoxemia results from any combination of five mechanisms.
Hypoventilation. Hypoxemia from hypoventilation alone has an increased PaCO2 and a normal A-a O2 gradient. The additional CO2 displaces inhaled oxygen in the alveolus. However, the remaining alveolar oxygen diffuses and mixes normally into the arterial blood, displaying a normal A-a O2 gradient as long as there is no alveolar or interstitial disease.
Right-to-left shunt. Right-to-left shunting occurs when blood enters the systemic circulation without traversing ventilated lung. There is always a small degree of right-to-left shunting because of the direct left atrial return of deoxygenated blood from both the coronary veins and the bronchial arteries. Increased right-to-left shunting occurs in a variety of conditions, including congenital cardiac malformation and acquired pulmonary disorders (pulmonary consolidation, pulmonary atelectasis). Regardless of the specific cause of the right-to-left shunt, it is always associated with an increase in the A-a O2 gradient. A hallmark of significant right-to-left shunting is the failure of arterial oxygen levels to increase in response to supplemental oxygen. Although a small improvement may be observed with supplemental oxygen, hypoxemia is never fully eliminated because of the mixing of deoxygenated blood in the systemic circulation.
Ventilation-perfusion (V̇/Q̇) mismatch. Ideal pulmonary gas exchange depends on a balance of ventilation and perfusion. Any abnormality resulting in a regional alteration of either ventilation or perfusion can adversely affect pulmonary gas exchange, resulting in hypoxemia. There are many causes of V̇/Q̇ mismatch, including pulmonary emboli, pneumonia, asthma, chronic obstructive pulmonary disease, and even extrinsic vascular compression. Regardless of cause, hypoxemia from ventilation-perfusion mismatch is associated with an increased A-a O2 gradient, and hypoxemia improves with supplemental oxygen.
Diffusion impairment. Pulmonary gas exchange requires diffusion across the alveolar–blood barrier. Regardless of the specific cause of the diffusion impairment, the A-a O2 gradient is increased, and hypoxemia improves with supplemental oxygen.
Low inspired oxygen. Decreased ambient oxygen pressure results in hypoxemia. This is commonly seen at high altitude (including commercial air travel) or in nonobstructive asphyxia. The A-a O2 gradient is normal, and hypoxemia improves with supplemental oxygen. For example, Denver, at 5400 ft (1646 m) above sea level, has an atmospheric pressure of 620 mm Hg and an inhaled PO2 of only 0.21 × 620 = 130 mm Hg, as opposed to 160 mm Hg at sea level.
There are three distinct acute compensatory mechanisms for hypoxemia. Initially, minute ventilation increases. Next, pulmonary arterial vasoconstriction decreases perfusion to hypoxic alveoli. Although vasoconstriction balances ventilation and perfusion to restore arterial oxygenation, it may also cause acute right heart failure and is ineffective with diffuse lung disease. Finally, sympathetic tone increases and improves oxygen delivery by increasing cardiac output, usually with an increased heart rate. Chronic compensatory mechanisms include an increased red blood cell mass and decreased tissue oxygen demands. These compensatory mechanisms appear to be activated at different levels of hypoxemia among different individuals. However, the acute compensatory mechanisms are always activated when PaO2 reaches 60 mm Hg, and compensatory mechanisms fail when PaO2 falls below 20 mm Hg.
The signs and symptoms of hypoxemia are nonspecific. CNS manifestations include agitation, headache, somnolence, coma, and seizures. Although tachypnea and hyperventilation are often present, by the time PaO2 is <20 mm Hg, there is a central depression of respiration. Cyanosis, the blood or tissue discoloration associated with a lowered arterial oxygenation saturation, is not a sensitive or specific indicator of hypoxemia. Patients with chronic compensatory mechanisms may display polycythemia or alterations in body habitus (e.g., pulmonary cachexia).
Hypoxemia is defined as a PaO2 <60 mm Hg, so formal diagnosis requires arterial blood gas analysis. Although pulse oximetry is useful for screening purposes and decreased SaO2 readings accurately predict significant hypoxemia, clinically acceptable pulse oximetry saturation readings (>90%) do not exclude hypoxemia. If certain hemoglobin abnormalities exist (e.g., methemoglobin or carboxyhemoglobin), pulse oximetry analysis may overestimate oxygen saturation and under measure the response to supplemental oxygen (see the section “Cyanosis,” below). Regardless of the specific cause of hypoxemia, the initial approach remains the same: ensuring a patent airway and providing supplemental oxygenation with a goal of maintaining a PaO2 >60 mm Hg. Except in patients with right-to-left shunts, arterial oxygenation responds to supplemental oxygen.
HYPERCAPNIA
Hypercapnia is exclusively caused by alveolar hypoventilation and is defined as a PaCO2 >45 mm Hg. Alveolar hypoventilation has many causes, including rapid shallow breathing, small tidal volumes, underventilation of the lung, or reduced respiratory drive. Hypercapnia never results from increased CO2 production alone (Table 62-3).
Depressed central respiratory drive Structural CNS disease: brainstem lesions Drug depression of respiratory center: opioids, sedatives, anesthetics Endogenous toxins: tetanus Thoracic cage disorders Kyphoscoliosis Morbid obesity Neuromuscular impairment Neuromuscular disease: myasthenia gravis, Guillain-Barré syndrome Neuromuscular toxin: organophosphate poisoning, botulism Intrinsic lung disease associated with increased dead space Chronic obstructive pulmonary disease Upper airway obstruction |
A portion of each tidal volume remains in the non–gas-exchange portion of the respiratory system—termed the dead space—that is determined by the anatomic size of the conducting airways (trachea and bronchi). The portion of the tidal volume that reaches the alveoli is that which remains after the dead space volume is subtracted:
Alveolar ventilation (VA) per minute is the alveolar volume multiplied by the respiratory rate:
Alveolar hypoventilation can result from a decrease in respiratory rate, a decrease in VT, or an increase in dead space. Dead space volume can increase above that due to the anatomic size of the conducting airways, such as seen as a result of ventilation of lung portions with deficient or absent perfusion; these ventilated portions do not participate fully in gas exchange because of inadequate blood flow.
Medullary chemoreceptors stimulate both respiratory rate and VT in response to increased CO2, so that alveolar ventilation is finely controlled relative to CO2 production and PaCO2 is maintained within a narrow range. Decreased respiratory drive is associated with CNS lesions and toxic depression (Table 62-3). Thoracic cage and neuromuscular disorders produce hypoventilation by slowing respiratory rate and/or decreasing VT relative to the production of CO2. Intrinsic lung diseases, such as chronic obstructive pulmonary disease, produce alveolar hypoventilation because of an increase in dead space.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


