Fig. 22.1
Schematic of nephron. CCD cortical collecting duct, MCD medullary collecting duct (Adapted from [2])
The kidney receives approximately 20–25% of cardiac output. Blood enters the kidney from the aorta via the renal arteries, which divide into interlobar arteries. Each of these interlobar arteries travels through areas in which the renal cortex invaginate into the medulla between the medullary pyramids. Each interlobar artery is further divided into arcuate arteries, which travel parallel to the base of the renal pyramids at the junction of the renal cortex and medulla. Again, further division occurs into interlobular arteries that keep dividing until they become an afferent arteriole supplying a single nephron, which begins with the specialized capillary bed of the glomerulus. Blood exits the glomerulus via an efferent arteriole, before entering the peritubular capillary system. Drainage of the peritubular capillary system occurs via a small venue with venous drainage flowing back along a parallel path to the arterial system before finally emptying into a single renal vein and the vena cava. Of note the left renal vein is longer than the right renal vein. This anatomical difference does have clinical implications. For example, donor nephrectomies can be done laparoscopically if the left kidney is being harvested because of this difference, whereas an open procedure must be performed for right donor nephrectomy.
22.1.2 The Glomerulus
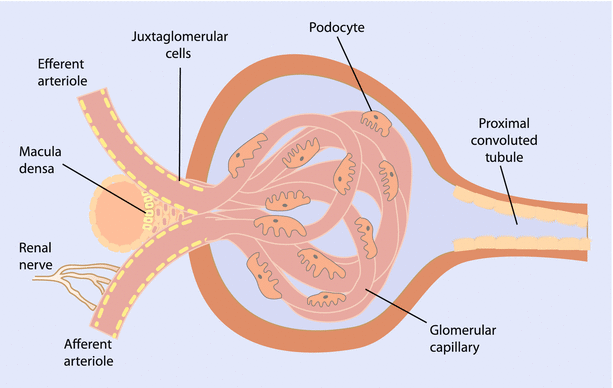
Filtration begins in the glomerular capillary bed. This specialized capillary bed resides in a space called Bowman’s capsule. The glomerular filter itself is made of 3 component parts: the endothelial cells of the glomerular capillary, the glomerular basement membrane, and specialized cells called podocytes or visceral epithelial cells (◘ Fig. 22.2) [3, 4]. The endothelial cells are different from those in other capillary beds in the body. They have fenestrations of roughly 70–100 nm in diameter that serve as a “size” filter. The glomerular basement membrane is a extracellular matrix of proteins created by fusion of the endothelial cell and podocyte basement membranes.
Podocytes provide support to the glomerular capillary complex and also form another slit type of filtration barrier by weaving together the little foot-like processes that extend from one cell to another. These foot-like processes support the glomerular capillary by wrapping around them and weave together with the foot processes of adjacent podocytes. The areas between the adjacent “feet” form the slit filtration barrier that makes up the third component of the overall glomerular filtration barrier. Healthy podocytes are very important for normal functioning of the glomerulus. Unfortunately, they have a limited ability to repair or regenerate themselves.
The glomerular barrier allows water to flow through freely, but acts as a barrier for particles based on both size and charge. Flow of albumin and anionic particles is restricted while small neutral particles and cations traverse the barrier freely.
Mesangial cells are another important cell type located in Bowman’s capsule (◘ Fig. 22.3) [4, 5]. These cells play a number of important roles in the glomerular apparatus. One of the primary roles they serve is in providing a support structure for the glomerular capillary network. They form a central tuft within the glomerulus and secrete different types of collagen, fibronectin, and other compounds that form a mesangial extracellular matrix. Mesangial cells help to support the structure of the glomerular loops and help to provide protection from high hydrostatic pressures. The mesangial cells have contractile properties that aid them in this task. Although the major regulation of blood flow to the glomerulus occurs as a result of changes in the efferent and afferent arterioles, the mesangial cells are thought to have the ability to fine tune intraglomerular blood flow. The extracellular matrix produced by glomerular cells also have important immunologic and homeostatic functions. Production of free radicals, cytokines, and chemokines all occur when these cells become activated. Mesangial cells appear to “talk” with podocytes and endothelial cells through mechanisms that are still being investigated [6]. These interactions prove to be important during disease processes as dysfunction in one cell type can result in alterations in the others.
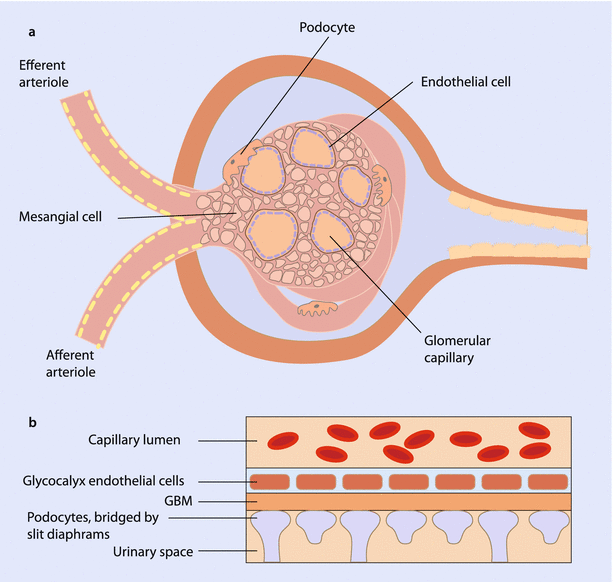
Fig. 22.3
a Cross section of glomerulus illustrating relationship between mesangial cells, podocytes, and endothelial cells. b Representation of the 3-part filtration barrier comprised of the podocytes, glomerular basement membrane (GBM), and endothelial cells (Adapted from [5])
Two classic glomerular diseases that are associated with mesangial cell function are immunoglobulin A (IgA) nephropathy and diabetic nephropathy. In IgA nephropathy, mesangial cells are injured by the deposition of IgA, which results in mesangial cell proliferation and increased production of the mesangial matrix components through the work of cytokines and growth factors. In turn this activation appears to result in changes in podocyte function leading to protienuria. Patients who develop diabetic nephropathy demonstrate pathologic changes in mesangial cells as well, as demonstrated by expansion of the mesangial matrix, mesangial cell hypertrophy and proliferation, and the development of inflammation. These changes contribute to the global renal dysfunction seen in this condition.
What determines the rate of flow through the glomerulus? Within the glomerulus, the amount of filtered fluid will depend on the counterbalancing forces of hydrostatic pressure and oncotic pressure. Thus at the beginning of the glomerular capillary, the hydraulic force of fluid will overcome the oncotic pressures working to keep fluid within the capillary bed. As fluid exits the capillary, protein concentration and thus oncotic pressure will increase until the two forces balance and no further net filtration of fluid occurs. Another factor affecting flow is represented by ultrafiltration coefficient, Kf. This value represents factors such as the surface area available for filtration, and the “leakiness” of the capillary filter. One final component affecting the glomerular filtration rate (GFR) will depend on the rate of flow of fluid through the glomerular capillaries. When fluid is flowing slowly through the glomerulus, the point of equilibrium between the hydrostatic forces driving fluid out into Bowman’s capsule and the oncotic pressure created by plasma proteins may occur earlier along the capillary path. Thus, for the remaining portion of the capillary bed left to traverse, no net filtration occurs. When fluid travels more quickly it takes more time for this point of equilibrium to occur. Thus, although there may be less net filtration at one single point, the longer path and thus greater time that filtration occurs leads to a net increase in the amount of filtered fluid. Differing resistances in the afferent and efferent arterioles modulate this rate of flow depending on hormonal factors, drugs, and other vasoactive substances.
22.1.3 Proximal Tubule
Once the fluid filtered by the glomerulus exits Bowman’s capsule it enters the proximal tubule, which is the major site of reabsorption. Through this process of reabsorption the proximal tubule regulates the extracellular fluid volume of the body, reclaims important electrolytes and nutrients, and regulates acid base balance [7]. These proximal tubule cells can be thought of as bulk processing centers. They reabsorb the majority of the substances that were filtered, with the fine tuning of many processes accomplished further downstream of this component of the nephron.
To understand the manner in which proximal tubule cells accomplish all of the aforementioned tasks, we must first understand the general makeup of the tubular cells in the nephron. Each cell type, whether they be in the proximal tubule or in the collecting duct, is oriented with two interfaces: one to the tubular fluid, and the other with blood from peritubular capillaries. The membrane lying at the interface with the tubular fluid is referred to as the apical membrane, while the basolateral membrane refers to the blood-cell interface. Many cellular processes in the nephron are powered by a protein transporter complex known as the Na+-K+-ATPase either directly or indirectly. The Na+-K+-ATPase uses the energy achieved from ATP hydrolysis to transport three molecules of sodium out of the cell while moving two molecules of potassium into the cell’s interior. This mechanism is referred to as “active transport.” In most cells along the nephron this process occurs along the basolateral membrane. The net effect of this process results in the establishment of a concentration gradient such that the interior of the cell has a lower sodium concentration and higher potassium concentration compared to the exterior. The combination of the ratio of three positive cations removed from the cell for the only two imported, as well as subsequent conductance of potassium back out of the cell, results in the establishment of a negative intracellular voltage. The energy potential created by this concentration gradient and voltage difference is then leveraged to transport many solutes. Examples of the use of this strategy recur repeatedly along subsequent areas in the nephron. In the following sections, we will discuss the individual mechanisms the proximal tubule is thought to use to accomplish the reabsorption of NaCl, water, bicarbonate, amino acids, phosphates and important metabolites. The proximal tubule is also involved in the excretion of drugs and endogenous toxins, but this will be addressed in the section on renal drug excretion.
NaCl, Water, and Glucose Reabsorption
The electrochemical gradient, with the low intracellular Na+ levels and negative intracellular lumen, powers the transport of both sodium chloride and water into the cell at the apical membrane. This is accomplished by multiple different exchangers as there is no transporter protein that reabsorbs NaCl as a single unit. Instead import of Na+ is tied to organic compounds such as glucose, H+, and sulfates. In a sense, Na+ “catches a ride” with whatever transporter protein that can accommodate it. Chloride anion transport occurs through transporters tied to organic bases and other anions at the apical boarder and via a variety of transporters at the basolateral membrane. Also a significant amount of chloride is reabsorbed via the paracellular pathway. In this route, chloride rides down the concentration gradient in passive transport between the proximal tubule cells. The net effect of all these processes results in the reabsorption of 60–70% of the filtered NaCl and water as well as reabsorption of >99% of filtered glucose by the time the end of the proximal tubule is reached [7, 8]. Because water and solutes are reabsorbed together, the reabsorption occurring in the proximal tubule is generally isotonic in nature.
Regulation of the HCO3 − Buffer
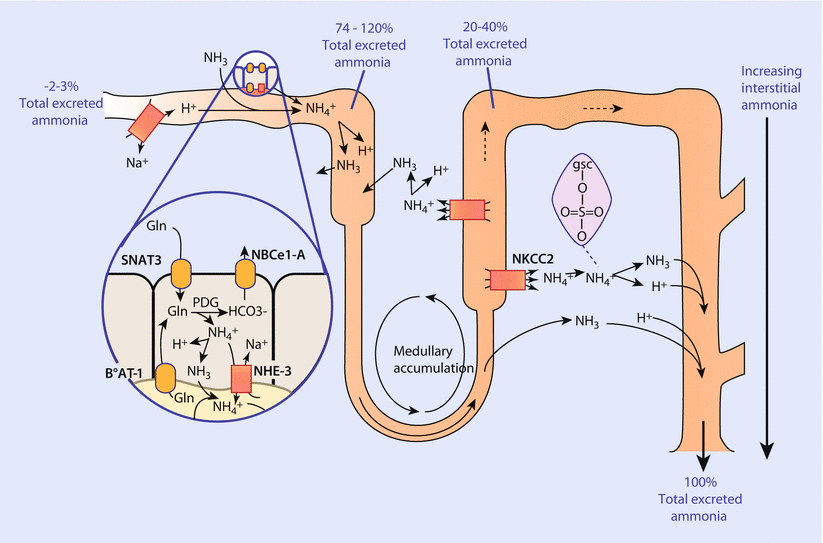
Bicarbonate represents one of the most important buffers in the human body. Variation in the levels of bicarbonate contribute to the maintenance of the acid base status, which needs to be regulated within a very precise range. The proximal tubule plays a major role in the regulation of bicarbonate, reclaiming 70–90% of filtered bicarbonate. Surprisingly, this is not accomplished by simply transporting sodium bicarbonate from the tubular lumen across the apical membrane. Instead, the proximal tubule secretes hydrogen ion into the tubular lumen through the work of NHE3, a protein that functions as an electroneutral Na+/H+ exchanger. Hydrogen ion is moved into the tubular lumen as the Na+ is transported into the cell (◘ Fig. 22.4).
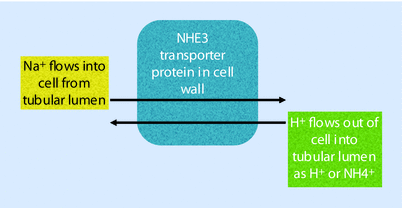
Fig. 22.4
Regulation of the HCO3 − buffer
This hydrogen anion can then follow 1 of 2 basic paths: (1) titrate another substrate that acts as an acid carrier that is ultimately excreted in the urine, or (2) undergo transformation into a form that leads to the reclamation and regeneration of bicarbonate. Together these 2 processes are referred to as “renal acidification” [7].
NHE3 (the Na+/H+ exchanger protein) not only secretes H+ but also secretes the acid NH4 + directly into the tubular lumen. The path of renal acidification through titration of carriers that reside in the tubular lumen, mentioned previously, include compounds such as ammonium and the divalent form of hydrogen phosphate. When ammonium combines with the hydrogen ion, NH4 + is generated in the tubular lumen:
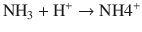
When hydrogen ion titrates phosphate to its monovalent form it functions as an acid that is secreted in the urine, much like the ammonium cation.
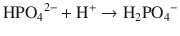
The second path of bicarbonate reabsorption can be accomplished a couple of different ways as well. The enzyme carbonic anhydrase catalyzes the combination of bicarbonate and hydrogen ion to form carbon dioxide, a substance that can easily diffuse across the apical cell membrane:
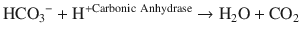
Once inside the cell, carbonic anhydrase regenerates both the hydrogen ion and bicarbonate. A transporter protein on the basolateral membrane called NBC1 then transports the bicarbonate into the peritubular capillary along with Na+, thus reclaiming bicarbonate.
Alternatively, the secreted hydrogen ion can combine with citrate. The change in charge of citrate after its combination with hydrogen makes it compatible with a protein cotransporter called NaDC-1 (Na+ decarboxylate cotransporter) that imports the citrate along with Na+ ion into the cell. Citrate is then ultimately metabolized in the proximal tubule cell (in 1 of 2 manners) both resulting in the net generation of bicarbonate, which again can be transported across the basolateral membrane by NBC1. Thus, in summary, the hydrogen ion that was initially transported out of the proximal tubule cell combines with citrate allowing it to be transported from the tubular lumen into and across the proximal tubular cell where it is ultimately reabsorbed into the blood. ◘ Figure 22.5 portrays a nice summarization of these pathways and the transporter proteins involved [7].
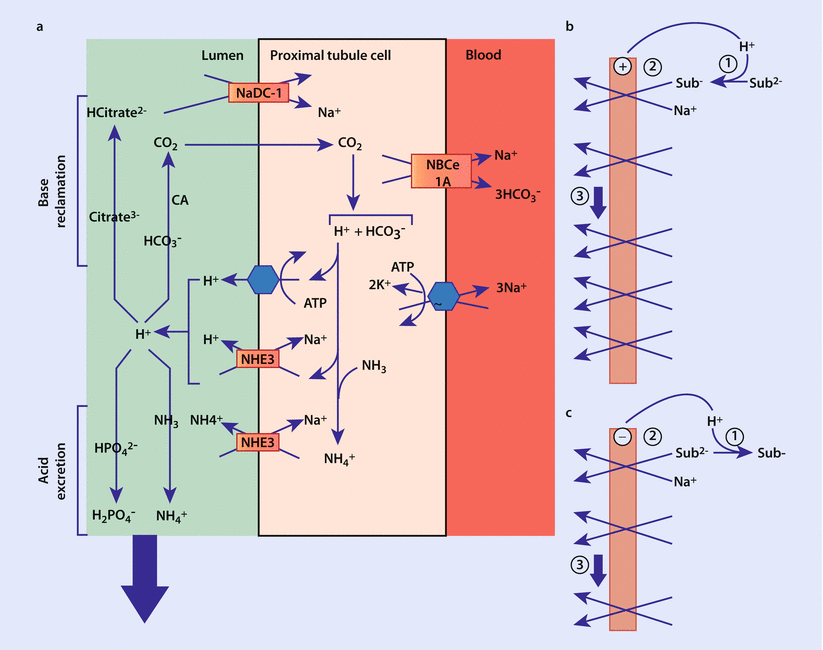
Fig. 22.5
Illustration of proximal tubule management of filtered bicarbonate. a Carbonic anhydrase catalyzes the luminal conversion of bicarbonate into CO2 and water. The CO2 diffuses through the cellular membrane where it again is converted by carbonic anhydrase into hydrogen ion and bicarbonate. Hydrogen ion can be secreted back into the tubular lumen by either the H+-ATPase, or more commonly the Na+/H+ exchanger NHE3. NHE3 also has the ability to transport NH4 +, which is generated intracellularly from NH3 and H+. Once hydrogen ion is secreted into the lumen it may be either recycled to participate again in the reabsorption of bicarbonate; may convert citrate to its bivalent form, which is transported into the cell; or may be used to titrate urinary acids, which are excreted. The degree to which the proximal tubule can reabsorb bicarbonate and acidify urine can be either unregulated or down regulated as illustrated in b and c, respectively. Sub – substrate (Adapted from [7])
Transport of Amino Acids
The majority of amino acids filtered by the glomerulus are neutrally charged, and easily reabsorbed by a specific transporter protein at the apical membrane. Separate transporters exist for acidic and basic amino acids. One important amino acid to be familiar with is glutamine due to the role it plays in the renal response to metabolic acidosis [7]. Under normal physiologic conditions only a small amount of glutamine undergoes uptake (approximately 20%) and metabolism (<3%). However, this picture rapidly changes under the acute onset of metabolic acidosis. During the onset of acute metabolic acidosis, the amount of glutamine that the kidneys are exposed to increases as a result of release from muscle tissue. The proximal tubule cell reabsorbs a greater amount of glutamine (roughly 35%) from the tubular lumen and also takes up glutamine from the peritubular capillary blood. This glutamine is then metabolized to generate greater amounts of ammonium cations (which are excreted in the urine), glucose, and bicarbonate (which undergoes reabsorption) (◘ Fig. 22.6) [7]. When metabolic acidosis becomes more chronic, the circulating levels of glutamine drop back to a level that is 70% of normal. However, the percentage of glutamine extracted remains elevated due to continued extraction from both membranes, a process which is facilitated through the increased expression of certain transporter proteins. The enzymes involved in glutamine metabolism have an increased expression resulting in an higher capacity for ammonium cation and bicarbonate production. The expression of proteins such as NHE3 (Na+/H+ exchanger protein) and NBC1 (protein that moves bicarbonate across the basolateral membrane of the cell) is increased as well, augmenting the acidification of urine and reabsorption of bicarbonate.
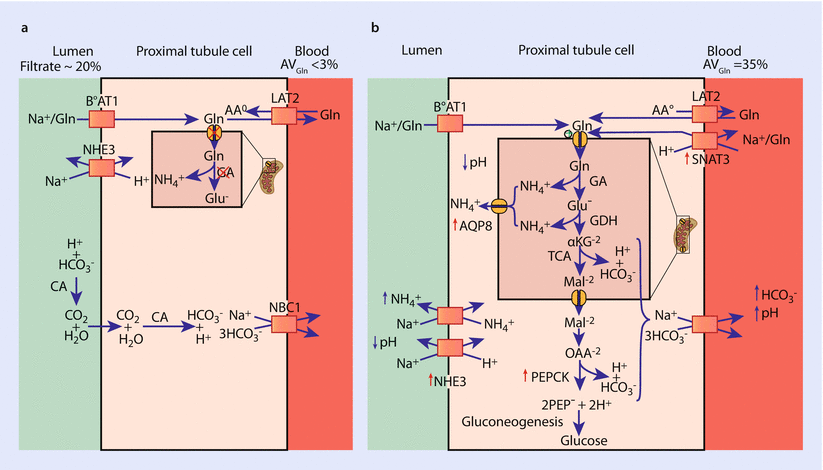
Fig. 22.6
Glutamine metabolism. a Represents handling of glutamine in a normal acid – base environment. b Illustrates the catabolism of glutamine during conditions of chronic acidosis demonstrating its catabolism to form bicarbonate, which is reabsorbed into the blood, NH4+ which acidifies the urine, and glucose (Adapted from [7])
Phosphate
The renal component of phosphate balance is managed almost exclusively by the proximal tubule [7, 9]. Again the electrochemical gradient established by the Na+-K+-ATPase serves as the driving force for the apical reabsorption of phosphate via three separate transporters. The mechanism by which phosphate exits the basolateral membrane and is reabsorbed into the blood is not well understood. The amount of phosphate reabsorbed versus lost in the urine will depend on dietary intake as well as the input of signaling factors from parathyroid hormone, dopamine, fibroblast growth factor 23, and a transmembrane protein called klotho.
22.1.4 Thin Limbs of the Loop of Henle
Once it exits the proximal tubule, the path of tubular fluid traverses through the descending limb of the loop of Henle (LoH) down into the renal medulla. After traveling through a hairpin curve, it then ascends back up until it reaches the thick ascending limb (TAL) of the loop of Henle. The region from the corticomedullary junction to the papilla is characterized by an increasing osmolarity gradient that plays a critical role in the ability of the body to produce concentrated urine. In fact the osmolarity from the outer renal cortex to the inner renal medulla from values of 300 mOsm to as high as 2700 mOsms. The mechanism by which gradient is established and maintained is not entirely understood, and older models put forth have not accommodated data measured in experimental models. Thus, the understanding of this process continues to evolve [10].
The best current understanding of how the kidney establishes the osmolar gradient begins with an acknowledgement of the special characteristic of the descending and ascending thin loops themselves (◘ Fig. 22.7) [10]. The first important characteristic relates to water permeability [2]. Contrary to prior models, it has been demonstrated that the descending thin limbs (DTLs) of short looped nephrons fail to express the transmembrane protein aquaporin-1 (AQP1). AQP1, aquaporin, is a water channel that makes the tubule water permeable. For long-looped nephrons, the descending thin limbs only express AQP1 in the initial 40% of their length. Thus, most of the descending thin limbs are actually impermeable to water. Ascending thin limbs (ATLs) are water impermeable as well. While these segments have long stretches of impermeability to water, they do have a high degree of permeability to sodium chloride beginning from just before the hairpin bend and continuing on through the ascending thin limb. Finally, both the descending and ascending loops of Henle have a high permeability to urea along their entire lengths.
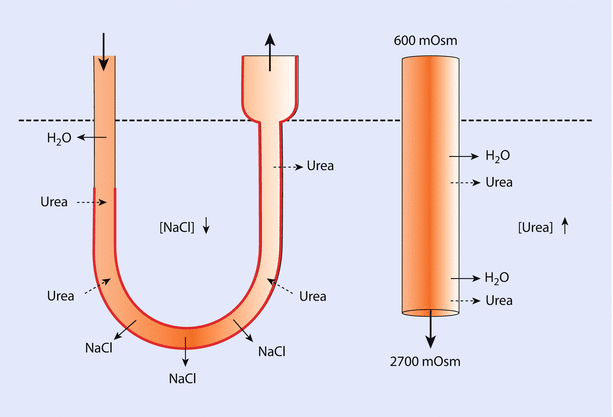
Fig. 22.7
Thin limb of loop of Henle. This figure demonstrates the current understanding of the role of water permeability and urea in creating the hypertonic environment that is responsible for the ability to concentrate urine. The thick red line demonstrates the portion of the thin limb that does not express aquaporin and is thus not permeable to water. In contrast, permeability of urea exists along the length of the tubule. Around the bend of the loop, NaCl is passively reabsorbed (Adapted from [10])
As tubular fluid flows down the descending thin limb, water exits the DTL lumen until it reaches the area in which AQP1 expression ceases, at which point water egress ceases. As the DTL dives into the renal medulla, the osmolarity of the tissue surrounding it increases due to increases in interstitial urea concentration. Thus, urea diffuses down its concentration gradient from its high concentration into the tubular fluid. As the bend of the LoH is approached, NaCl passively exits the cell, but due to the lack of water permeability there is no concurrent egress of water. At the bend of the LoH, the tubular fluid is surrounded by the highest osmolar gradients it will see until it again descends back through the inner medulla via the collecting ducts. As the fluid exits the bend and rises back up toward the renal cortex, the concentration of urea begins to decrease the closer the fluid travels to the cortex. Urea will continue to travel down its concentration gradient. Thus, in the areas of the ascending limb that are deep, urea will diffuse into the tubule. However, at some point the fluid in the tubule will have a urea concentration that is greater than the surrounding intersitium, resulting in its diffusion back out into the interstitium. This process is referred to as “counter current multipication” [10].
22.1.5 Thick Ascending Limb of Loop of Henle
The mechanism described previously depends on a high osmolar gradient with a high concentration of urea and low concentration of NaCl in the inner medulla. How does this gradient become established and maintained? In order to understand this process we must continue on to understand the workings of the thick ascending limb (TAL) and collecting ducts (CD) (◘ Fig. 22.8) [2]. The TAL is impermeable to water and travels back up to the renal cortex where the cells of the macula densa abut the afferent and efferent arterioles of the glomerulus. The macula densa represents specialized cells in the TAL that play a role in tubuloglomerular feedback. While movement of NaCl occurs in a passive fashion in the thin limbs, it is actively transported out of the lumen in the TAL. The net result is an increasingly dilute luminal fluid that contributes to counter current multiplication.
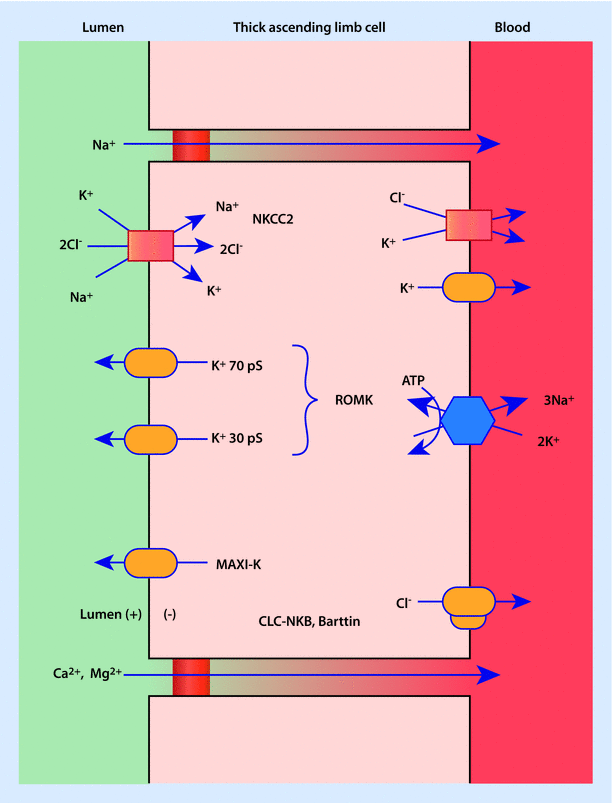
Transport of NaCl is mediated by the protein complex NKCC2 at the apical membrane [2]. NKCC2 cotransports Na+, K+, and 2Cl− to the intracellular space. This transport protein is of clinical importance because it is extremely sensitive to furosemide. Furosemide inhibits the transport of Cl− across the cell and appears to have effects on the cells of the macula densa, which also contains NKCC2. In the macula densa, Lasix (furosemide) functions to inhibit tubuloglomerular feedback and suppress renin released by chloride in the tubular lumen.
Potassium plays a very important role in the TAL [2]. Its presence is necessary for the NKCC2 (furosemide sensitive, electroneutral Na+-K+-2Cl− cotransporter) transporter to function. Since there is very little potassium in the tubular fluid at this point, in order for continued active transport of NaCl to occur, then the potassium must be recycled back across the apical lumen so that it may be reused. Renal outer medullary potassium channel (ROMK) and maxi-K (also known as BK or “big” K) potassium channels at the membrane fulfill this task. The majority of the potassium transported across the apical membrane is recycled back into the tubular lumen, whereas the NaCl is transported across the basolateral cell membrane courtesy of the Na+-K+-ATPase and chloride channels. Thus, the active transport of NaCl in the TAL represents an example of secondary active transport. This process of NaCl reabsorption and K+ recycling creates a positive potential difference in the tubular lumen.
Other functions of the TAL include reabsorption of more cations via the paracellular pathway [2]. This includes additional sodium, roughly 55% of filtered magnesium, and around 20% of filtered calcium. Part of the impetus for this movement is found in the positive-lumen potential difference established by furosemide-sensitive, electroneutral Na+-K+-2Cl− cotransporter (NKCC2), and is aided by channels in the tight junctions between cells that have an affinity for cations.
Additionally, the TAL contributes to maintenance of acid-base balance through the reabsorption of approximately 15% of filtered bicarbonate [2]. The mechanism by which this is accomplished involves electroneutral Na+/H+ exchanger (NHE3) as described earlier in the proximal tubule. The TAL also is important in the body’s management of ammonia. When the ammonia produced by the proximal tubule reaches the TAL, a large percentage of it is reabsorbed back into the systemic circulation. This reabsorbed ammonium anion is transported to the liver where it undergoes metabolism to form urea. The reabsorption of ammonium anion is carried out mechanistically by apical transport of ammonium anion from furosemide-sensitive, electroneutral Na+-K+-2Cl− cotransporter NKCC2 (although NH4 + appears to be able to be transported by any transporter protein that handles K+) and another protein transporter at the basolateral membrane called NHE4 (another electroneutral Na+/H+ exchanger). The capacity of the TAL to reabsorb ammonium anion increases under conditions of acidosis. Interestingly, NH4 + can be reabsorbed via the paracellular pathway as well. Ultimately, the transport of ammonium anion by the TAL establishes another gradient contributing to counter current multiplication, with the highest amounts of ammonia in the inner medulla and lowest levels located in the renal cortex.
The body’s ability to adjust urine production and composition to meet metabolic needs depends on hormonal regulation [2]. Various hormones target the TAL to regulate ion transport. Substances such as vasopressin, parathyroid hormone, glucagon, calcitonin, and B adrenergic activation all increase ion transport through an increase in cyclic adenosine monophosphate (cAMP) levels. As an example, interaction of vasopressin with V2 receptors leads to an increased activity in electroneutral Na+-K+-2Cl− cotransporter (NKCC2) activity as well as an increase in the number of these proteins on the apical membrane. With prolonged exposure to vasopressin, the TAL cells hypertrophy and double their baseline activity of NaCl transport. In contrast, prostaglandin E2, extracellular calcium, and nitric oxide negatively influence ion transport.
22.1.6 Distal Convoluted Tubule
Once tubular fluid passes the macula densa it enters the distal convoluted tubule (DCT) [11]. Tubular fluid is now very dilute as a result of the active transport of NaCl and impermeability of the TAL. The measured transepithelial voltage, or the voltage difference between the DCT cells and the tubular lumen, remains very close to zero at the beginning of the DCT. The continued reabsorption of NaCl (described later) results in the formation of a negative charge in the tubular fluid. This negative voltage in turn helps to drive the secretion of K+, along with the reabsorption of chloride, calcium, and magnesium.
The DCT is comprised of 2 distinct segments: the early or DCT1 segment, and the late, or DCT2 segment (◘ Fig. 22.9) [11]. These segments differ from one another in a number of ways. The first difference relates to the ability of the mineralocorticoid receptor (MR) to be stimulated by steroid hormones. In the DCT1 region, the MR responds to both glucocorticoids and mineralocorticoids. However, expression of an enzyme called 11-B hydroxysteroid dehydrogenase 2 in the DCT2 section prevents cortisol from exerting its effects at the mineralocorticoid receptor. Thus, the DCT2 segment is much more sensitive to the effects of aldosterone than the early DCT. A second difference relates to the manner in which NaCl reabsorption is executed. In the early DCT, sodium transport at the apical membrane occurs in an electro-neutral manner. The transporter protein NCC (thiazide-sensitive NaCl cotransporter), transports both one Na+ cation and one Cl− anion together simultaneously. Activity of NCC also happens to be inhibited by thiazide diuretics. The coupled movement of sodium chloride maintains the zero voltage potential in the tubular lumen. In the late DCT, NCC continues to be expressed and function, but another transmembrane protein called the epithelial sodium channel (ENaC) also contributes to sodium reabsorption. The key difference lies in the electrogenic nature of this transport. Since there is no concomitant transfer of an anion to balance out the movement of the positively charged Na+ in the epithelial sodium channel (ENaC), a voltage difference begins to occur and the tubular lumen develops a negative potential. The impetus for this influx of Na+ at the apical membrane can again be traced back to our old friend the Na+-K+-ATPase functioning at the basolateral membrane. Remember, 3 Na+ are transported out for every 2K+ carried into the cell creating a negative, low [Na+], high [K+] intracellular environment. The “leak” of potassium back across the basolateral membrane by various potassium channels recycles the potassium and allows this electrochemical gradient to be regenerated and maintained. Basolateral chloride transport is mediated by a chloride-specific channel (ClC-Kb) as well as a KCl transporter protein (KCC4).
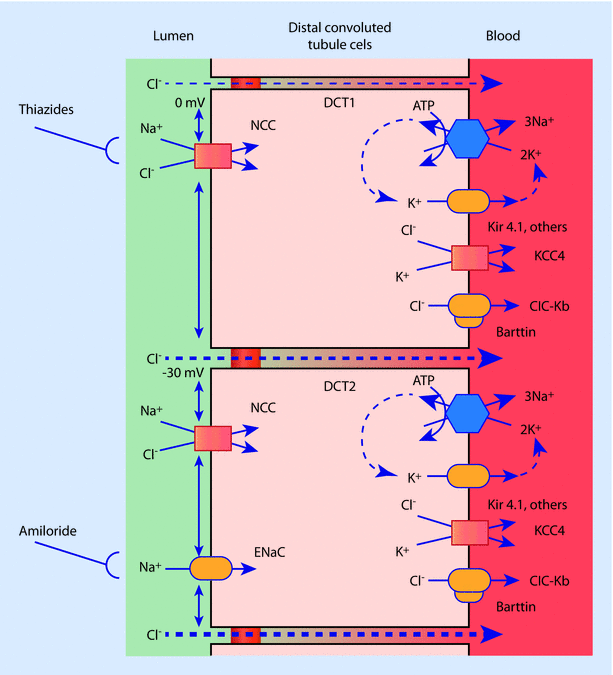
The voltage difference generated by the combined effects of an epithelial sodium channel (ENaC) and the Na+-K+-ATPase helps drive the movement of other solutes. Paracellular movement of chloride is one example. Other interesting examples are found in the active transport of calcium and magnesium, which will not be expanded upon here. Perhaps the most interesting example lies in the handling of potassium at the apical membrane. When tubular fluid exits the DCT2 it contains more potassium than was originally present due to secretion across the apical membrane [11]. The secretion of potassium increases the further down one travels in the DCT in proportion to the increasingly negative voltage potential. The faster the fluid travels down this path, the more potassium is secreted as well. These effects are mediated by 2 different potassium channels, which we have discussed previously: the renal outer medullary potassium channel (ROMK) and BK (“big” K or maxi K). In the TAL, the function of these proteins at the apical membrane is to recycle potassium back into the tubular lumen to allow furosemide-sensitive, electroneutral Na+-K+-2Cl− cotransporters (NKCC2) to initiate the reabsorption of NaCl. In the DCT, however, their function at the apical membrane is to secrete potassium into the tubular lumen. Renal outer medullary potassium channel (ROMK) is voltage sensitive and it secretes more potassium as the voltage differential becomes more negative. When tubular fluid flows at an increased rate then the BK (“big” K) channel becomes activated due to the sheer stress. Intracellular calcium and nitric oxide are postulated to play a role in this. Thus, high flow rates and a high sodium load will activate both ROMK and BK channels leading to enhanced potassium secretion.
One final interesting fact about ROMK relates to the role that magnesium plays in the regulation of this transporter [11]. When magnesium binds to the renal outer medullary potassium channel (ROMK) on the intracellular side of the transport protein, it blocks the secretion of potassium. Thus, a lack of sufficient magnesium levels will lead to a more “open” ROMK channel and increased loss of potassium through the urine resulting in hypokalemia. This hypokalemia will not be corrected by simply administering exogenous potassium chloride. Instead, until the magnesium deficiency is corrected, the hypokalemic state will persist.
Knowledge of the receptors and mechanisms involved in sodium reabsorption in the nephron may seem overly detailed, but it does have implications on patient care and clinical practice. Diuretics play a very important role in the management of many patient populations, and medications such as furosemide are administered routinely. With continued administration of furosemide its effectiveness wanes. The mechanism behind the phenomenon can be easily understood. NKCC2 (furosemide-sensitive, electroneutral Na+-K+-2Cl− cotransporter) sensitivity to furosemide leads to inhibition of NaCl reabsorption in the TAL, thereby increasing the sodium load delivered to NCC (thiazide-sensitive NaCl cotransporter) transporters in the DCT. Rates of flow in the tubular fluid will increase, as will the electrogenic reabsorption of sodium. As a result of the increasingly negative voltage and flow, ROMK and BK (both potassium transport channels) will increase potassium secretion. The hypokalemia that is observed with furosemide administration is thus explained [11].
If a patient is on Lasix chronically, the DCT1 segment adapts through cellular hypertrophy in order to increase the capacity to reabsorb sodium [11]. Remember, the distal convoluted tubule 1 segment’s ability to reabsorb sodium occurs only as a result of the work of the thiazide-sensitive NaCl cotransporter. Hypertrophy of the cell leads to an increase in the number of the thiazide-sensitive NaCl cotransporters (NCC). This leads to an ability to increase the amount of sodium reabsorbed. This process mitigates the effectiveness of the diuretic with furosemide thereby leading to diuretic resistance. However, since the action of NCC (the thiazide-sensitive NaCl cotransporter) may be inhibited by thiazide diuretics, then their administration may circumvent this adaptive response. In effect, the administration of the thiazide diuretic knocks out the ability of the kidney to reabsorb the increased sodium load that was delivered to the distal convoluted tubule from the administration of furosemide.
22.1.7 Collecting Ducts
Before tubular fluid exits the nephron it must finally pass through the collecting ducts (CD). The cells in the collecting ducts make the final set of fine-tuned adjustments in the areas of water balance, acid base homeostasis, and inorganic solutes such as sodium, chloride, and potassium [12, 13]. Two cells types—principal cells and intercalated cells—carry out the responsibilities of this nephron segment and are interspersed with one another along the length of the epithelium.
The principal cell focuses its efforts in two main areas: sodium chloride and water reabsorption (◘ Fig. 22.10) [13]. The electrochemical gradient that acts as the driving force for sodium reabsorption is once again created by the Na+-K+-ATPase at the basolateral membrane. However, the ENaC protein channel acts as the vessel through which entrance occurs at the apical membrane [12]. Thus, in the CD, the sodium is not actively transported. The transport is electrogenic in nature, and contributes to the secretion of potassium through ROMK and hydrogen ion in a manner similar to what was described in the late DCT. The ENaC receptor is regulated by a number of hormones including aldosterone, atrial natriuretic peptide, and arginine vasopressin. After stimulating the mineralcorticoid receptor, aldosterone multiplies the number of receptors at the apical membrane, thereby magnifying its effects. Insulin also stimulates ENaC in the principle cell. Conversely, ANP inhibits the ENaC channel in addition to repressing secretion of renin and production of aldosterone.
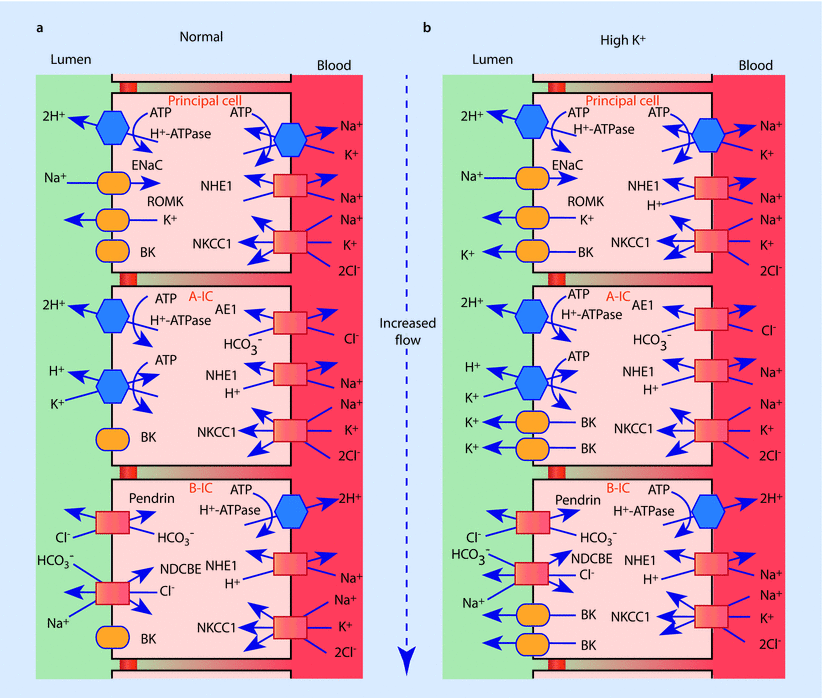
Fig. 22.10
Collecting duct principal cell, type A and type B intercalated cell. Illustration of receptors in each cell type as well as variability of potassium secretion depending on tubular flow. a Normal potassium secretion. b Potassium secretion as affected by high tubular flow (Adapted from [13])
Principal cells mediate water reabsorption through the aquaporin channels [12]. AQP2 is expressed at the apical membrane while AQP3 and AQP4 are located at the basolateral membrane. By this point in the nephron approximately 90% of the water has already been reclaimed. The amount of fluid reabsorbed at this point is highly dependent on vasopressin levels. Vasopressin is able to modulate the water permeability of the collecting duct though its effects on AQP2. With vasopressin stimulation, AQP2 stores in the cell’s interior are transferred to the apical membrane. Additionally, vasopressin can alter protein expression of AQP2 for a longer term effect. Dopamine and prostaglandin E2 antagonize these effects by initiating a mechanism that returns AQP2 from the apical membrane to back to intracellular vesicles.
Intercalated cells (IC) make up the remaining population of the collecting duct [13]. One key difference between intercalated cells and almost every other cell described in this chapter lies in the power source driving transport events. Instead of the Na+-K+-ATPase, an H+-ATPase serves as the generator of the electrochemical gradient in intercalated cells. Similar to cells within the distal convoluted tubule, intercalated cells are very rich in mitochondria and all IC contain the enzyme carbonic anhydrase. Three subtypes of intercalated cells exist: type A, type B, and non-A non-B. Type A intercalated cells help to acidify urine. Type B and non-A non-B cells secrete bicarbonate into the tubular lumen.
Type A intercalated cells position the H+-ATPase at the apical lumen. In a combined effort with a H+-K+-ATPase pump, protons are secreted into the lumen [13]. The loss of the proton drives intracellular carbonic anhydrase to generate bicarbonate, which is reabsorbed by the transporter protein AE1 across the basolateral membrane in exchange for a chloride anion. A clinical example of the importance of this mechanism is found in the inherited forms of distal renal tubular acidosis. In these patient populations, the normal functioning of type A intercalated cells is upset, resulting in a lack of ability to acidify urine. This disruption leads to a state of acidemia, alkalization of urine, and often nephrolithiasis.
Potassium balance can be fine-tuned through the effects of type A IC. The cells can participate in both potassium reabsorption and potassium secretion depending on body conditions. The H+-K+-ATPase pump at the apical membrane creates a mechanism for the reabsorption of K+ from the lumen [13]. However, in response to high luminal flow, a negative voltage gradient, or increased intracellular calcium potassium can be secreted through the BK (maxi-K) channel as has been previously discussed [14].
In type B intercalated cells the H+-ATPase pump is positioned at the basolateral membrane [13]. The apical membrane contains a transporter protein named pendrin, which excretes a bicarbonate anion into the lumen in exchange for a chloride anion. The presence of an electro-neutral Na+/HCO3 −/Cl− transporter allows these cells to play a role in NaCl reabsorption as well. Thus the two roles of the type B intercalated cells include volume expansion and bicarbonate secretion.
The final topic that needs to be addressed relates to the pivotal role that the collecting duct plays in the generation of the urea gradient necessary to drive counter current multiplication and the body’s urine concentrating ability. In the previous discussion of the LoH we discussed the manner in which urea underwent a passive diffusion down its concentration gradient into the tubular lumen from the renal interstitum. What we neglected to mention, however, was genesis of the high concentration gradient in the renal interstitium. A number of urea transporters in the collecting duct establish and help to maintain the high concentration of urea in the inner medullary intersitium [15]. Located at the apical membrane, the urea transporter UT-A1 carries urea from the tubular lumen into the collecting duct cell. The urea is then transferred across the basolateral membrane by the urea transporter UT-A3. The activity and number of these transporters is regulated by vasopressin. In conditions of high osmolarity, or volume depletion, vasopressin increases the number and activity of the urea transporters thereby increasing the concentration of urea in the inner medullary intersitium. The increased osmolarity allows for increased reabsorption of water and further concentration of urine.
What prevents the concentration gradient established in the inner medulla from being diluted by the blood flowing down the vasa recta? Counter current exchange of urea preserves the gradient. The descending vasa recta and red blood cells contain the urea transporter protein UT-B1. This urea transporter allows the red blood cells to absorb urea as they descend into the inner renal medulla and effectively matches the osmolarity of the blood cells with that of the surrounding interstitium. Thus, no water flows out to dilute the surrounding tissue. As the red cells and vasa recta ascend back toward the renal cortex, UT-B1 helps the red cell to quickly eject urea back into the interstitium. This facilitated transport decreases the osmolarity of the red cells, prevents urea from being carried out of the inner medulla, and reinforces the medullary concentration gradient.
22.2 Integrated Processes in the Nephron
The aforementioned journey through all the sections of the nephron has demonstrated some of the cellular mechanisms and processing techniques that the kidney uses in order to adapt to the myriad of internal and external changes the body is confronted with (◘ Fig. 22.11) [8, 9, 14–16]. ◘ Tables 22.1 and 22.2 summarize key functions, characteristics, and receptors of each nephron segment. ◘ Table 22.3 delineates the handling of key electrolytes and solutes in the glomerular filtrate as they pass through the nephron. Now we will use this newfound understanding to broaden our perspective from the microscopic world of the single nephron in order to examine more global processes such as the defense of the body’s fluid volume and acid-base status.