where Q is the net fluid filtration from the pulmonary microvascular (intravascular) capillary system to the peri-microvascular (extravascular) pulmonary tissue, Kf is the capillary filtration coefficient, P is the hydrostatic pressure, given as Pmv and Ppmv in the microvascular and peri-microvascular compartments, respectively, δ is the protein reflection coefficient – varying between 0 (freely permeable) and 1 (non-permeable to proteins) – and π is the protein colloid osmotic pressure in the microvascular (πc) and peri-microvascular (πt) tissue. The expression Kf reflects the surface area available for fluid and the hydraulic conductance for fluid.
It is obvious from this expression that edema may form in a tissue for a variety of reasons or combination of factors. In the lung, we recognize HPE as opposed to HPPE. The idea here is that the former may be a result of mainly extrapulmonary factors, such as left heart failure. In contrast, the latter type of edema is mainly caused by an alteration of the permeability of the capillary membranes in the lung, permitting fluid and large molecules to extravasate even with normal vascular pressures. Naturally, the therapeutic approach, apart from symptomatic support, differs according to the etiology of edema formation.
Experimental studies on pulmonary edema
Edema may accumulate because of a combination of altered hydrostatic and permeability factors. This can, for example, be seen in early experimental septicemia caused by injecting live bacteria or lipopolysaccharides. Sheep typically react with both a sharp increase in pulmonary artery pressure and an increased permeability in lung capillaries.[9] Both these factors, therefore, contribute to edema formation in the lung.
To what degree the maintenance of a high airway pressure is detrimental has been the subject of considerable debate. We have experienced an era where the idea of “super PEEP” was advocated by some groups in the treatment of ALI. Because MV was introduced in the treatment of ALI, it was obvious that barotrauma, with or without pneumothorax, was an adverse effect to consider when using intermittent positive-pressure ventilation.[10] The Kolobow group studied sheep treated with MV that had their lungs overexpanded by the application of peak airway pressures at 50 cmH2O.[11] With this ventilator setting, the clinical picture of ARDS could be produced by MV alone.
Webb and Tierney created impressive alveolar edema in rats by ventilating with high airway pressures for only 20 min.[12] This work was confirmed and extended by Dreyfuss and Saumon [13], suggesting the term “volotrauma” as opposed to “barotrauma.” This latter study offered evidence that overexpansion of the lung tissue by volume alterations is more damaging than overexpansion by pressure alterations. We can conclude that severe lung damage with edema can be caused by airway-mediated mechanical factors, resulting in repeated tearing of tissue due to overexpansion by volume. West and Mathieu-Costello have published impressive electron microscopic evidence of how “overexpansion by pressure release” of either the airway or the capillary vessels (increased blood pressure) will result in “stress failure” of these structures.[14] The damaged structures allow fluid, larger molecules, and sometimes whole blood corpuscular elements to exit the circulation and participate in edema formation.
Our group measured lung lymph flow in anesthetized dogs ventilated with or without PEEP. During MV with PEEP 10 cmH2O, the lymphatic flow from the lung was reduced by almost 50% compared with that occurring with zero end-expiratory pressure. On the other hand, during SB, lung lymph flow was markedly elevated (Figure 13.1).[7] This indicates that intra-pulmonary pressure can influence lung fluid balance, and that SB considered in relation to lung lymph flow might have an advantage when compared with MV.
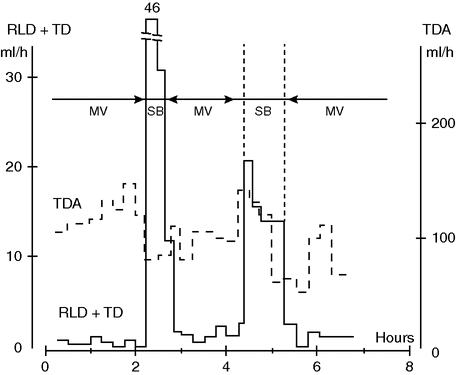
Pulmonary lymph flow (RLD + TD) and abdominal lymph flow (TDA) during mechanical ventilation (MV) and spontaneous breathing (SB) in a 30 kg anesthetized dog. Note the considerable increase in lung lymph flow during SB, suggestive of impeded lymph drainage during MV. Note also the decrease in abdominal lymph flow during SB. Whether this reflects decreased capillary leakage remains to be shown.
Summary of factors that promote pulmonary edema
A number of factors contribute to edema formation in the early stages of lung injury:
1. Increased capillary pressure (fluid overloading, left heart failure);
2. Elevated airway pressure causing barotrauma (MV, PEEP, and need for higher tidal volumes due to intra-pulmonary shunting);
3. Elevated airway pressure impeding lymphatic drainage of lung tissue and possibly pleural cavities;
4. Increased permeability of the capillary or alveolo-capillary membranes;
5. Impeded active transport of water from the alveoli back to the interstitium and circulation;
6. Extravasation of activated neutrophils and macrophages, which further lowers permeability after releasing inflammatory mediators locally and into the bloodstream;
7. Microthrombotization of lung blood or lymph capillaries by local activation of platelets, reducing the functional area of the lung capillary bed, which contributes to an elevated microvascular (hydrostatic) pressure (flow limitation) and possibly impedes the lymphatic flow.
Clearance of pulmonary edema
When discussing clearance of extravasated fluid from the lung, the situation becomes very complex. From a clinical point of view, it seems advisable on the basis of the above discussion to keep vascular pressures low (pulmonary artery pressure and central venous pressure). We attempted to optimize Starling factors by using a hyperoncotic, hypertonic infusion during pulmonary edema in dogs, but found to our surprise that this manipulation did not increase the rate of reduction of EVLW.[15] Obviously, other factors limited the maximum rate of all relevant factors that determine lung fluid balance.
An illustration of the need to take additional factors into account is the demonstration that the pleural cavities may act as safety factors delaying and minimizing edema formation. It is now known that a significant portion of interstitial fluid during edema formation is excreted into the pleural cavities and cleared from the parietal pleura.[16] Experiments on dogs demonstrate to what extent this animal is dependent on an intact lymphatic system to clear the pleural cavity of protein-rich, extravasated fluids. This was shown by ligating the lymphatic vessels draining the pleural cavity and measuring the rate of removal of indicator-labeled protein from a pleural effusion.[17] The dog could not remove such an effusion after lymphatic ligation. Our conclusion must be that a number of factors concerning clearance of edema, and safety factors against edema formation, have to be taken into consideration when discussing the fluid balance of lung tissue. We have previously suggested the calculation of “net fluid leakage” per time unit as a more relevant parameter to use for such an analysis.[18]
Summary of factors that facilitate the clearance of pulmonary edema
Our understanding today is that there exist several ways by which the lung can rid itself of edema. Fluid can be:
1. Locally reabsorbed through the capillary membrane by Starling-type mechanisms, or actively pumped out of the alveoli;[19]
2. Cleared through the lymphatic system as lymph and returned to the circulation;[8]
3. Excreted to the pleural space directly from the lung tissue and from there taken up into parietal pleural lymphatics for return to the circulation;[16]
4. Transported to the lung hilus interstitially and from there migrate into the mediastinum and be absorbed;[20]
5. Cleared through the airway.
Different pathways of fluid transport in the lung are shown schematically in Figure 13.2.[21]
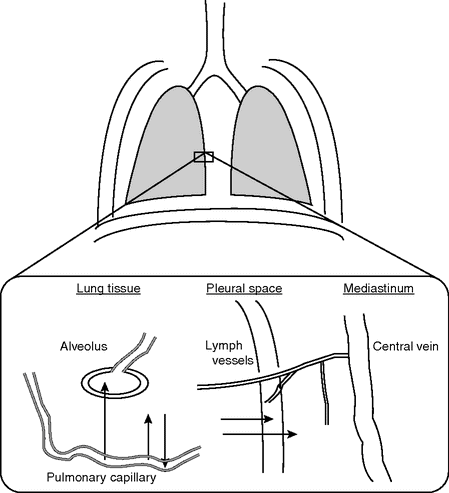
Pathways of filtered fluid from the intravascular to the extravascular spaces in the lung.
Ventilator-induced lung injury and pulmonary edema
A concept that has emerged during the past few years is lung damage caused by the ventilator per se, so called ventilator-induced lung injury (VILI). Positron emission tomography (PET) studies have shown that measures of barrier function are a nonspecific index of lung injury indicating functional, not structural, lung injury.[22,23] PET imaging methods can measure the rate at which proteins move across the endothelial barrier, from vascular to extravascular compartments, the so-called pulmonary transcapillary escape rate (PTCER). Palazzo et al. [24] used PET imaging to measure PTCER in an in vivo canine model of unilateral pulmonary ischemia-reperfusion injury and found it to be increased in the ischemic lung. Interestingly, both lungs had an increased PTCER when compared with control non-ischemic lungs, suggesting that injury in one lung can lead to similar injury in the contralateral lung, a finding that has been observed in an analogous clinical setting such as acute unilateral pneumonia.
Calandrino et al. described that, while PTCER and extravascular density (a close correlate to EVLW) were both elevated in patients with ARDS, they correlated poorly with one another on a regional basis.[22] Moreover, even as extravascular density returned to normal, PTCER remained elevated suggesting that lung tissue injury might be “subclinical” but still present, even after pulmonary edema has actually resolved. This was further confirmed by Sandiford et al. [25] who examined the regional distribution of PTCER and extravascular density more closely in ARDS patients and found ventral–dorsal gradients only for extravascular density but not for PTCER. Once more, functional injury was detected even in lung regions that appeared to be free of structural injury.
The finding that the lungs of patients with ARDS are more diffusely involved than what might otherwise be assumed from just structural radiological imaging, such as computed tomography, helps explain why ARDS lungs are so vulnerable to VILI: radiographically “normal” lung, i.e. lung with a normal EVLW content, in non-dependent lung regions may still be abnormal and vulnerable to mechanical stresses caused by MV. These data tell us that non-dependent regions of ARDS lungs are “at risk” because they demonstrate subclinical evidence of injury, which can be made manifest by inappropriate ventilator use. Jones et al. [26] evidenced a surprisingly high pulmonary uptake of [18F]-fluorodeoxyglucose (18F-FDG) in patients with head injury, at risk of developing ARDS but without lung symptoms at the time of the scan. This signal may reflect sequestration of primed neutrophils in lung capillaries. In vitro studies in isolated human neutrophils have demonstrated that the uptake of deoxyglucose is increased to the same extent in cells that are only primed, or primed and stimulated.[27] This indicates the vulnerability of these patients while on ventilatory support, because, even though neutrophils remain in a primed state, any additional stimulus precipitates actual tissue damage.
PET with 18F-FDG, which is a glucose-analog tracer, offers the opportunity to study regional lung inflammation in vivo. 18F-FDG is taken up predominantly by metabolically active cells and has been recognized as a key marker of neutrophilic inflammation in the inflamed non-tumoral lung.[28,29] There is a theoretical concern that in ALI 18F-FDG might leak to the alveolar spaces, becoming a major and non-specific determinant of the 18F-FDG signal, and potentially causing a false, non-inflammation-related increase in the measured uptake (Ki). The presence of edema, however, does not seem to affect the measurement of Ki, as suggested by Chen et al. [30] who measured glucose uptake with 18F-FDG in anesthetized dogs after intravenous oleic acid-induced ALI or after low-dose intravenous endotoxin followed by oleic acid. The rate of 18F-FDG uptake was significantly elevated in both endotoxin-treated groups, but not in the group treated only with oleic acid, leading the authors to conclude that pulmonary vascular leak, and consequently edema, does not significantly contribute to the Ki in ARDS lungs.
We studied the location and magnitude of early inflammatory changes using PET imaging of 18F-FDG in a porcine experimental model of ARDS.[31] We evaluated the individual contributions of regional injurious mechanisms during early stages of VILI. We found the highest uptake in the normal and poorly aerated regions, while the hyperinflated and non-aerated regions were similar to the control group (Figure 13.3). These findings challenge the current notion that hyperinflation and/or repeated collapse and re-expansion of alveolar units play the major role in early VILI. Instead, our data suggest that tidal stretch was highest in the poorly and normally aerated regions, and that this mechanism is the most important trigger of inflammation in these conditions. They also support the concept that the smaller the ventilated lung, the higher the VILI-triggering forces will be, as a larger fraction of tidal volume VT is delivered to a smaller lung volume.
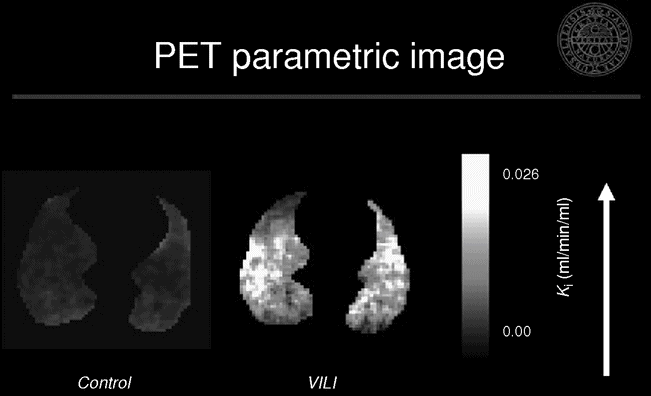
Representative positron emission tomography and X-ray computed tomography images of net [18F]-fluoro-2-deoxy-D-glucose uptake rate in an experimental model of ventilator-induced lung injury. Note the predominance of the activity in the normal and poorly aerated regions as seen in the corresponding X-ray computed tomography image.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


