Fig. 6.1
Barbituric acid ring (Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2016. All Rights Reserved)
Table 6.1
Structure activity relationship
Substitution | Effect | |
|---|---|---|
Position 5 | Aryl or alkyl group | Hypnotic and sedative effect |
Position 5 | Phenyl group | Anti-convulsant effect |
Position 2 | Sulfur | Rapid onset of action (thiopental) |
Position 1 | Methyl or ethyl group | Rapid onset of action (methohexital) |
Both sodium thiopental (◘ Fig. 6.2) and methohexital (◘ Fig. 6.3) are supplied for clinical use as a racemic mixture, prepared as sodium salts.
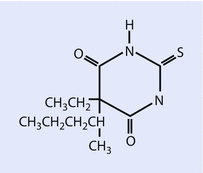
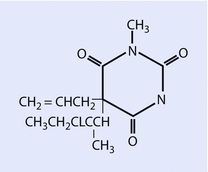
Fig. 6.2
Thiopental (Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2016. All Rights Reserved)
Fig. 6.3
Methohexital (Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2016. All Rights Reserved)
They are supplied as powder that is reconstituted with sterile water or normal saline. The powder is mixed with 6% anhydrous sodium carbonate to increase the water solubility.
The recommended solution is 2.5% for sodium thiopental and 1% for methohexital. These solutions are alkaline; pH 10.5 for sodium thiopental and 11 for methohexital. They can precipitate if mixed with acidic drugs such as rocuronium, atracurium, and vecuronium. It is recommended to flush the intravenous line before the addition of an acidic drug.
Sodium thiopental prepared under a sterile hood remains sterile and chemically stable for 6 days at room temperature and longer if refrigerated. A reliable potency of prepared methohexital exists 6 weeks after preparation at room temperature and its pH (10–11) inhibits any bacterial contamination.
6.2.2 Mechanism of Action
Gamma amino butyric acid (GABA) is the most abundant inhibitory neurotransmitter in the central nervous system (CNS).
GABAA receptors (◘ Fig. 6.4) are believed to mediate the hypnotic effect of most intravenous anesthetics as well as depression of the spinal cord reflexes and amnesia.
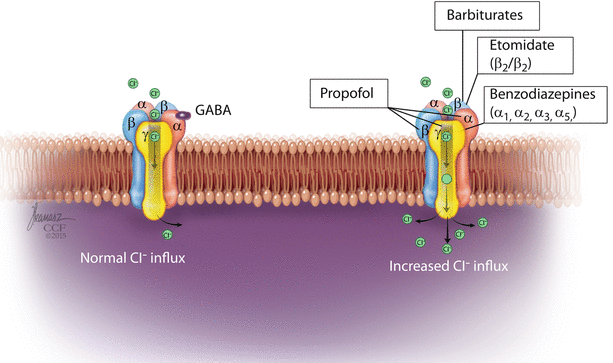
Fig. 6.4
GABAA receptor (Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2016. All Rights Reserved)
GABAA is a pentameric transmembrane glycoprotein receptor. It is a ligand-gated ion channel; when activated, transmembrane chloride conductance increases, hyperpolarizing the postsynaptic cell membrane and inhibits the function of the postsynaptic neurone.
Barbiturates act through modulating the action of GABAA receptors increasing the sensitivity to GABA. In high concentrations, barbiturates act as a direct agonist on GABAA receptors. On the GABAA receptor, barbiturates bind to a specific site on the B subunit.
It is believed that barbiturates inhibit neuronal nicotinic cholinergic receptors where thiopental acts as a competitive inhibitor. The clinical value of this action is unclear.
6.2.3 Pharmacokinetics of Barbiturates
Thiopental is alkaline, pH 10.5, but once it enters the circulation, it is buffered to the physiologic pH. It is highly lipid-soluble and this is the reason for its rapid onset of action that is within one circulation time. It is highly bound to albumin 75–90%. Protein binging is affected by the concentration of plasma proteins, displacement by other drugs, and changes in the blood pH where alkalemia decreases the protein binding. Hypoalbuminemia necessitates decreasing the induction dose and consequently the pharmacologically active free drug.
Redistribution out of the CNS is the main mechanism for termination of the anesthetic effect after injection of a single bolus dose. It takes 3–4 redistribution half-lives for termination of effect. The redistribution half-life (t1/2α[alpha]) is 2–4 min. Termination of the pharmacological effect is prolonged when the drug is given in repeated doses or as an infusion.
Subsequent metabolism and elimination is much slower with a half-life (t1/2β[beta]) of >10 h. Thiopental is metabolized mainly in the liver. Thiopental undergoes ring desulfuration to pentobarbital that is an active metabolite with a longer duration of action and represents around 3.5% of the given dose, but this percent can increase with high doses.
Both pentobarbital and thiopental undergo oxidation to pharmacologically inactive metabolites. Oxidation is the most important pathway for thiopental metabolism. Hydroxylation also takes place to inactive metabolites. Thiopental inactive metabolites are excreted in urine.
Elderly patients show a slower rate of distribution, which warrants adjusting the dose of thiopental. Conversely, recovery time after large or repeated doses is faster in infants than adults due to accelerated elimination. Patients with poor cardiovascular reserve show an exaggerated response to the hypotensive effect of thiopental.
Zero order kinetics (a constant fraction of the drug regardless of the total dose of the drug) describes the metabolism of thiopental after repeated doses or continuous infusion. Chronic administration of barbiturates or other drugs that induce the oxidative microsomal enzymes enhances the metabolism of barbiturates.
Methohexital is metabolized by the liver to inactive hydroxylated metabolites. It has a faster clearance than thiopental and accumulates less after prolonged infusions and this can be explained by the hepatic extraction ratio of methohexital (0.5), which means that the liver extracts 50% of the drug reaching it while the hepatic extraction ratio for thiopental is 0.15.
6.2.4 Pharmacodynamics of Barbiturates
Cardiovascular System
Barbiturates cause direct myocardial depression and peripheral vasodilatation that is more pronounced on the capacitance vessels. They inhibit the medullary vasomotor center, which results in a decrease in the arterial blood pressure that is exaggerated with rapid injection, in hypovolemic patients, and the elderly. The depressive effect on the sympathetic nervous system is more than that on the parasympathetic, and this can result in bradycardia. In fact, tachycardia is more observed and attributed to the effect of hypotension on the baroreceptors.
The decrease in blood pressure with induction is more related to venodilatation than direct myocardial depression.
Central Nervous System
Barbiturates exert a dose-dependent, progressive depression of the central nervous system that involves the spinal reflexes. Thiopental suppresses the electroencephalogram (EEG) in a progressive fashion to burst suppression and electrical silence. Methohexital can increase the seizure activity in epileptic patients, which makes it suitable for induction of anesthesia in patients coming for electroconvulsive therapy (ECT). Thiopental is an excellent anticonvulsant, it terminates seizure activities fast, but status epilepticus might require repeated doses or continuous infusion.
Barbiturates decrease the cerebral metabolic oxygen consumption rate (CMRO2), the cerebral blood flow (CBF), cerebral blood volume (CBV), and intracranial pressure (ICP). They are potent cerebral vasoconstrictors and might have a protective effect against focal ischemia (inverse-steal phenomenon).
Thiopental lacks analgesic effect and even has an antanalgesic effect in sub-anesthetic doses.
Respiratory System
Barbiturates are respiratory depressants; they depress the medullary respiratory center and decrease the sensitivity to hypoxia and hypercapnia. The decrease in minute ventilation is mainly due to decrease in tidal volume.
Laryngeal spasm is noticed with barbiturates more than other sedative-hypnotics. Histamine release is described with thiopental and the increase in bronchial smooth muscle tone might be attributed to it.
Hepato-Renal
There is no significant effect on the hepato-renal function with a single induction dose of barbiturates. Barbiturates are liver microsomal enzymes inducers and can increase the metabolism of other drugs.
Effect on Other Organs
Barbiturates have no direct neuromuscular junction effect but can decrease the skeletal muscle tone in high doses. During methohexital induction, muscle tremors, myoclonus, and hiccups are noticed.
Thiopental crosses the placenta and can depress the newborn and its activity, but it lacks a significant effect on the uterus.
Intraocular pressure decreases 25–40% with thiopental.
6.2.5 Uses of Barbiturates
Induction of Anesthesia: A dose of 4–6 mg/kg of thiopental induces general anesthesia in 30 s, the dose is reduced in the elderly, patients with poor myocardial function and hypovolemia, and the rate of injection is slowed to avoid an exaggerated hypotensive response. In an obese patient, the dose should be calculated on the lean body weight. A dose of 1–1.5 mg/kg induces general anesthesia with methohexital, accompanied by mild pain. Barbiturates are not used in maintenance of anesthesia due to prolonged recovery. Barbiturates are used to induce barbiturate coma after neurological insults.
Treatment of status epilepticus
Neuroprotection in elevated intracranial pressure and with focal ischemia.
6.2.6 Side Effects of Barbiturates
Hypotension
Allergic reactions, true allergic reactions are rare but direct histamine release is noticed.
Bronchospasm in asthmatic patients.
Laryngeal spasm.
Local irritation and tissue damage if injected in the subcutaneous tissues.
Intra-arterial injection can precipitate acute ischemia because of the severe arterial spasm due to release of endogenous vasoconstrictors, precipitation of thiopental crystals, intimal damage, and thrombosis.
On the occasion of intra-arterial injection, the catheter is left in place, a vasodilator (papaverene) is injected through the catheter, and a stellate ganglion block is used to promote vasodilatation and start heparin infusion.
Barbiturates can precipitate acute porphyria in patients with intermittent or variegate porphyria due to aminolevulinic acid synthetase (ALA-synthetase) induction with the accumulation of porphobilinogen during hemoglobin production. Acute attacks can be fatal. Patients present with severe abdominal pain, nausea, vomiting, and neurological abnormalities. For this reason barbiturates are contraindicated in patients with porphyria.
Sodium thiopental production was interrupted in the United States in 2010, and due to controversy over its usage in lethal injections, the European import was halted.
6.3 Etomidate
Etomidate is a carboxylated imidazole intravenous sedative-hypnotic that is available as a single enantiomer, the R(+) isomer that is 10–20 times more potent as a hypnotic than the S(−) isomer. It is poorly soluble in water with moderate lipid solubility. It is prepared as a clear solution in 35% propylene glycol at a concentration of 2 mg/ml or as a lipid emulsion. It has a pH of 6.9 so the concern of precipitation with acidic drugs as rocuronium is nonexistant.
6.3.1 Mechanism of Action
Etomidate carries out its action through modulation of the GABAA receptors and acting as a GABA agonist.
It shows high selectivity to the receptor with stereo-selectivity. It binds to receptors containing β(beta)2 and /or β(beta)3 subunits and less to receptors containing β(beta)1 subunits.
Αlpha2 adrenergic receptor activation is described and it may contribute to its hemodynamic effects where activation of the vascular smooth muscle α(alpha)-2b receptors causes vasoconstriction and hypertension.
6.3.2 Pharmacokinetics
Etomidate is 75–77% bound to albumin. It is metabolized by ester hydrolysis to inactive metabolites mainly in the liver. Excretion is mainly in urine and to a lesser extent in bile. Its elimination half-life is 1–5 h and if given as a continuous infusion its context-sensitive half time is shorter than propofol.
6.3.3 Pharmacodynamics
Cardiovascular System
The superiority of etomidate among intravenous induction anesthetics arises from its stable hemodynamic profile that results in minimal or no change in preload, heart rate, and myocardial contractility.
Central Nervous System
Etomidate has a rapid onset of unconsciousness and a short duration of action (2–4 min after an induction dose). It decreases the CMRO2, decreasing CBF and the ICP. Etomidate maintains or even increases the cerebral perfusion pressure (CPP) and it is a potent cerebral vasoconstrictor.
When used for induction of general anesthesia in electroconvulsive therapy (ECT), the seizure activity is longer than that achieved with methohexital. It may increase the EEG activity in epileptogenic foci but it still has an anti-convulsive effect.
Compared to propofol and barbiturates, it has less effect on the motor-evoked potentials.
Respiratory System
Etomidate has a minimal depressant effect on respiration but this can be pronounced if combined with other respiratory depressants as opioids. The ventilatory depressant effect of etomidate is not exaggerated in chronic obstructive pulmonary disease (COPD).
Endocrine System
Etomidate is a potent inhibitor of cortisol and aldosterone synthesis, even more potent than its hypnotic effect. Adrenocortical suppression is dose dependent and it is due to the inhibition of 11B-hydroxylase that catalyzes 11B-hydroxylation in cortisol synthesis and aldolase in aldosterone synthesis. It is believed that the imidazole structure is responsible for this adrenocortical suppression. A single induction dose of etomidate can inhibit the cortisol synthesis and the normal adrenocortical response up to 12 h. This can be prolonged more than 24 h after infusions. Prolonged infusion of etomidate in the critically ill ventilated patient has been shown to increase the mortality by two-fold.
There is still controversy on the impact of the effect of etomidate single dose and using it in critically ill patients, especially those with hemodynamic instability where etomidate usage is attractive.
6.3.4 Uses of Etomidate
The main usage for etomidate is for intravenous induction of anesthesia particularly in patients with compromised hemodynamics.
0.2–0.4 mg/kg for intravenous induction that needs to be modified in elderly patients with reduced protein binding.
6.5 mg/kg used for rectal induction of anesthesia.
When used as an infusion; 5 mcg/kg/min for sedation and 10 mcg/kg/min for maintenance of anesthesia where its context sensitive half-time is shorter than propofol.
6.3.5 Side Effects of Etomidate
Adrenocortical suppression (see earlier).
Nausea and vomiting. It is reported to be 30–40% in propylene glycol preparation but the same incidence of nausea as propofol in the lipid emulsion preparation.
Myoclonic activity and not convulsions is commonly seen with etomidate induction that can be minimized by premedication with opioid or benzodiazepine.
Pain on injection is more described if etomidate is injected in a small vein, especially with the propylene glycol preparation. This incidence of pain is less when preceded by lidocaine, injection in a big vein, or the lipid emulsion preparation is used. It is believed that this pain is due to activation of a member of the Transient Receptor Potential (TRP) ion channel family, TRPA1. TRPA1 is a principle receptor in nociception and together with TRPV1 contributes to neurogenic inflammation and pain signaling. Etomidate and propofol evoke pain through activation and sensitization of TRPA1.
Controversy exists about using etomidate in patients with porphyria and its safety. Some, reports its safe usage but it is an inducer for ALA-synthetase.
6.3.6 The Future of Etomidate Analogs
The attractive pharmacological profile of etomidate with its cardiorespiratory stability effects stimulated the research for modifying its structure to avoid its side effects. The preclinical trial of methoxycarbonyl-etomidate (moc-etomidate) describes an analog that is rapidly metabolized by non-specific esterases. With a half-life of a few minutes, even when it suppresses the adrenocortical function, it would be for a much shorter duration than the parent drug.
Carboetomidate is the other member of the future etomidate analogs. The imidazole ring of the parent drug that is responsible for the adrenocortical suppression is replaced by a pyrrole ring. This produces a drug that has the same hypnotic potency and cardiorespiratory effects of the parent drug but with less adrenal suppression.
6.4 Ketamine
Ketamine is an arylcyclohexylamine related to phencyclidine (PCP) in its structure. It is prepared as ketamine hydrochloride that is water soluble and ten times more lipid soluble than thiopentone. The preparation available in the United States is a racemic mixture of its S(+) and R(−) isomers. Some countries in Europe and Latin America have the preparation that only contains the S(+) enantiomer that is 3–4 times more potent, has a shorter duration of action, and less emergence reactions.
6.4.1 Mechanism of Action
The pharmacological effect of ketamine compared to barbiturates, propofol, and benzodiazepines is through the interaction with different receptors in the CNS. It is widely known to be a non-competitive antagonist of the N-methyl-D-aspartate (NMDA) receptor, it has antagonist effect on the cholinergic muscarinic receptors, agonistic effect on the α(alpha) and β(beta) adrenergic receptors, agonist for the opioid receptor, and it enhances the dopaminergic activity.
The NMDA receptor complex is a cation gated ion channel (mainly calcium and to a lesser extent potassium and sodium). When the receptor is activated by endogenous excitatory amino acid agonists (aspartic acid, glutamic acid, and glycine), the channel is opened, and the calcium influx increases causing depolarization of the membrane that plays an important role in the development of the sensory impulse at the cortical and subcortical levels.
Ketamine binds the phencyclidine (PCP) binding site within the ion channel in the open state preventing the influx of calcium and consequently preventing the membrane depolarization.
6.4.2 Pharmacokinetics of Ketamine
Ketamine is unique among intravenous induction agents in that it can be administered via different routes; it can be administered via intravenous, intramuscular, oral, nasal, and rectal route. It is water soluble, highly lipid soluble, 12% bound to plasma proteins with a pKa of 7.5.
Ketamine is rapidly absorbed; maximum plasma concentration is achieved in 5–15 min after intra-muscular (IM) injection, 20 min after nasal, and 30 min after oral administration.
It undergoes extensive first pass effect in the liver and intestines that is responsible for the low bioavailability of ketamine after oral and rectal administration (<30%).
It is the high lipid solubility and the low protein binding that allows ketamine to cross the blood brain barrier rapidly.
Termination of its pharmacological effect is through redistribution away from its CNS site of action. Redistribution half-life is 2.7 min.
Ketamine metabolism is mainly through N-demethylation to norketamine by hepatic cytochrome 450 enzymes. Norketamine is three-fourths the potency of the parent drug that undergoes hydroxylation, conjugation, and excretion mainly through the kidneys and to a lesser extent in feces. Ketamine elimination half-life is approximately 2 h. Oxidative metabolism of ketamine cyclohexanone ring is also described.
6.4.3 Pharmacodynamics of Ketamine
Cardiovascular System
Ketamine has a dual action, a direct cardiac depressing action that is seen in patients with exhausted catecholamines (critically ill and shocked patients), preventing the indirect effect of ketamine to take the upper hand. The indirect (predominant) effect is due to inhibiting the re-uptake of circulating catecholamines causing tachycardia, hypertension, increase in contractility, increase in cardiac output, increase in systemic vascular resistance, and increase in the pulmonary artery pressure. These hemodynamic changes can cause an imbalance between myocardial oxygen supply and demand, precipitating myocardial ischemia.
Central Nervous System
Ketamine induces a state of dissociative anesthesia (dissociation between the thalamo-neocortical and limbic system), where patients seem awake, with opened eyes, a nystagmus gaze and phonating, but dissociated from the environment with variable degrees of hypertonus and muscle movements. The anesthetic state is accompanied by intense analgesia. On emergence, patients have alterations in their mood, dysphoria, floatation sensations, vivid dreams, and hallucinations.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


