Fig. 14.1
Illustration of the coagulation cascade with its intrinsic (contact activation) and extrinsic (tissue factor) pathway. Each pathway generates a series of reactions in which inactive circulating enzymes and their co-factors are activated. The key elements of the extrinsic pathway involve the release of tissue factor from trauma or vascular injury. In the presence of calcium, tissue factor forms a complex with factor VIIa and cleaves clotting factors X and IX to their active forms. The intrinsic pathway is based on contact activation where factor IXa binds to factor VIIIa and forms intrinsic tenase, which activates factor X. The interaction of factor Xa with factor Va results in the formation of prothrombinase, which converts factor II to IIa (thrombin). Factor IIa further amplifies the coagulation system, converts soluble fibrinogen to insoluble fibrin, and also activates platelets. Anticoagulants are interacting at various stages with this coagulation system and interrupt the enzymatic reactions (Reprinted with permission from Alquwaizani et al. [1])
Perioperatively, hemostasis is often challenged but perturbations of the integrity of the vascular endothelium and subsequent platelet activation may result in acute clot formation in the arteries or veins, which ultimately manifests as arterial or venous thromboembolism. This is where anticoagulant drugs, including antiplatelet therapies and antithrombotic agents are utilized to interfere with hemostasis.
When selecting the most appropriate anticoagulant drug, thorough consideration of the efficacy-to-safety ratio is warranted. In addition, there are a number of pharmacological factors that affect a drug’s success: the absorption rate, the overall metabolism, an active parent drug rather than a prodrug that needs to be metabolized, drug interactions, interpatient variability in anticoagulant effects (e.g., drug, environment, and genetic), onset of action, pharmacokinetics with a dose-dependent drug effect, elimination, and a direct target. The overarching goal over the last several years has been to develop antithrombotic, anticoagulant, and antiplatelet drugs that carry more favorable pharmacological properties (e.g., faster onset of action, fewer interactions, and less interpatient variability in their antithrombotic effects) thus reducing thrombotic events without generation of unacceptably high bleeding rates. Besides more original drugs such as unfractionated heparin (UFH), the antiplatelet agents aspirin and clopidogrel and the vitamin K antagonist warfarin, modern treatment options now include anticoagulants that directly target factor IIa (bivalirudine, argatroban, dabigatran) or Xa (rivaroxaban, apixaban, edoxaban) and the next-generation antiplatelet drugs prasugrel and ticagrelor. This chapter focuses on the pharmacological properties of the most commonly used anticoagulants, antithrombotics, and antiplatelet drugs.
14.2 Unfractionated Heparin, Low-Molecular-Weight Heparin, and Fondaparinux
Unfractionated heparin (UFH) is the anticoagulant of choice in acute settings (such as cardiovascular procedures or surgery) while both UFH and low-molecular-weight heparin (LMWH) are preferred agents in acute thrombosis. Heparin is an endogenously produced, linear polysaccharide that consists of repeating units of pyranosyluronic acid and glucosamine residues. Heparins contain an active pentasaccharide sequence that binds to the endogenous serin protease inhibitor antithrombin (AT), thus causing a conformational change of AT, which results in accelerated AT binding and inactivation of coagulation factors XIIa, IXa, XIa, Xa, and thrombin. Neither UFH nor LMWH have significant intrinsic fibrinolytic activity and neither provides direct thrombolysis. Most important for the clinical effect of heparin is the accelerated inhibition of thrombin and factor Xa. Once a heparin molecule has formed a complex with AT, it can easily dissociate and bind to other AT molecules, thus providing a continuous anticoagulant effect. The active pentasaccharide sequence (e.g., high-affinity-material) responsible for catalyzing AT is more prevalent in the chains of UFH than LMWH and only 30–50% of the UFH- and <20% of the LMWH molecules contain the specific and active pentasaccharide sequence. Fondaparinux is a fully synthetic analog of the naturally occurring pentasaccharide found in heparins, which selectively and irreversibly binds to AT. This results in neutralization of factor Xa, which ultimately inhibits thrombin formation and thrombus development.
Unfractionated heparin is administered either intravenously (IV) or subcutaneously. As with other medications, intravenously administered UFH achieves therapeutic plasma concentrations and its clinical effect more quickly compared to subcutaneous administration. Owing to its low bioavailability, subcutaneous UFH needs to be dosed more generously (>30,000 U/day) to reach a clinical effect. Intravenously administered heparin can be effectively monitored and adjusted based on infusion rates. To monitor the antiocoagulant effect of UFH, the activated partial thromboplastin time (aPTT) or the anti-factor Xa essay (range 0.3–0.7 IU/mL) are being utilized.
The clearance of UFH from the systemic circulation is dose-dependent and follows 2 independent mechanisms. The initial clearance is rapid and depends on saturable binding of UFH to endothelial cells, macrophages, and local proteins where UFH undergoes depolymerization. The second phase depends on renal-mediated clearance, is non-saturable, and results in non-linear UFH pharmacokinetics with anticoagulation increasing disproportionally at high therapeutic doses. At clinically effective doses, UFH is primarily cleared via depolymerization. However, if clearance becomes dependent on the kidney function and especially if the renal function is impaired, prolonged administration of UFH results in a disproportionate increase in both the intensity and the duration of the anticoagulant effect. In patients with moderate (creatine clearance [CrCl] 30–50 mL/min) to severe renal dysfunction (CrCl <30 mL/min), over-anticoagulation occurs once saturation of reticuloendothelial clearance is reached. Hospital-specific weight-based dosing nomograms are useful to streamline the administration of UFH and have been associated with shorter time to therapeutic UFH levels and no increase in bleeding events. Major complications of UFH therapy include bleeding and heparin-induced thrombocytopenia (HIT). Heparin-induced thrombocytopenia (HIT) is a prothrombotic disorder caused by antibodies to macromolecular complexes of platelet factor 4 (PF4), a positively charged platelet protein, and heparin, a negatively chared carbohydrate. Diagnostic tests for HIT consist of the platelet count (decrease of >50% from baseline), immunoassays, and functional assays. Immunoassays identify antibodies against heparin/platelet factor 4 (PF4) complexes and use an enzyme-linked immunosorbent assay (ELISA). Functional assays such as the serotonin release assay measure the platelet-activating capacity of PF4/heparin-antibody complexes. While functional assays have greater specificity than immunoassays they are also time-consuming and not as widely available.
Intracranial, intraspinal, intraocular, mediastinal, or retroperitoneal bleeding is typically classified as major bleeding and usually results in hospitalization, transfusion, possible organ failure, and death. Risk factors for major bleeding during anticoagulation with UFH include the intensity of the anticoagulant effect, increased age, female sex, history of gastrointestinal bleeding, concomitant use of other anticoagulants, and duration of therapy. Patients receiving long-term heparin therapy are also at increased risk to suffer from osteoporosis and may be prone to vertebral fractures.
Perioperative anticoagulation management is procedure-dependent and informed by the patient’s risk factors for bleeding and thrombosis. Evidence-based guidelines for the perioperative management of antithrombotic therapy contain specific recommendations. Bridging therapy is recommended in patients at moderate to high risk of thromboembolism (e.g., the prevention of thromboembolism outweighs the potential for increased surgical bleeding). For patients at low risk for thromboembolism, no bridging therapy is recommended. Preoperatively, UFH is routinely halted for 4 h prior to the surgical procedure and the effect is monitored with a normalization of the aPTT. Postoperatively, therapeutic UFH is restarted after about 12 h but will be delayed if surgical bleeding continues. One key advantage of UFH anticoagulation is that its effect may be rapidly reversed with protamine. Each mg of protamine can neutralize 100 U of heparin. Importantly, protamine is associated with development of a number of adverse effects, ranging from life-threatening anaphylactic complications in 0.16% of patients to minor reactions associated with systemic hypotension in 13% of patients. The incidence and severity of the reaction to protamine may depend on the rate of administration (maximum recommended administration rate is 5 mg/min). Patients at risk for a reaction to protamine may be pretreated with corticosteroids and anti-histamine medications.
Evidence-based guidelines help to provide safe regional anesthesia in patients receiving antithrombotic or thrombolytic therapy. As long as patients receive only low-dose UFH subcutaneously, there is no contraindication for neuraxial techniques. However, in the patient that is receiving intraoperative (and possibly high-dose) anticoagulation with UFH, therapy should be started at least 1 h after the neuraxial needle placement. The removal of indwelling catheters such as epidural catheters should not be performed unless the UFH therapy has been halted for at least 2–4 h and not without demonstration of an effect on the level of coagulation (see ◘ Table 14.1).
Table 14.1
Pre- and postoperative recommendations for regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy as recommended by the American Society of Regional Anesthesia and Pain Medicine Evidence-Based Guidelines (Third Edition)
Preoperative |
Discontinue warfarin at least 5d before elective procedure |
Assess INR to 2d before surgery, if >1.5, consider 1–2 mg of oral vitamin K |
Reversal for urgent surgery/procedure, consider 2.5–5 mg of oral or intravenous vitamin K; for immediate reversal, consider fresh-frozen plasma |
Patients at high risk for thromboembolism |
Bridge with therapeutic subcutaneous LMWH (preferred) or intravenous UFH |
Last dose of preoperative LMWH administered 24 h before surgery, administer half of the daily dose |
Intravenous heparin discontinued 4 h before surgery |
No bridging necessary for patients at low risk for thromboembolism |
Postoperative |
Patients at low risk for thromboembolism |
Resume warfarin on postoperative day |
Patients at high risk for thromboembolism (who received preoperative bridging therapy) |
Minor surgical procedures – resume therapeutic LMWH 24 h postoperatively |
Major surgical procedures – resume therapeutic LMWH 48–72 h postoperatively or administer low-dose LMWH |
Assess bleeding risk and adequacy of hemostasis when considering timing of the resumption of LMWH or UFH therapy |
Recommendations from Douketis et al. [4] |
Not all invasive procedures/surgeries require normalization of the INR. |
Low molecular weight heparin or UFH is routinely administered as high-dose (therapeutic), intermediate-dose, and low-dose (prophylactic). Prophylactic-dose LMWH is commonly prescribed to prevent venous thromboembolism (VTE) but is not sufficient in the prevention of arterial thromboembolism (e.g., stroke in atrial fibrillation). Due to the homogeneity of the polysaccharide chains, LMWHs have a superior pharmacodynamic and pharmacokinetic profile in comparison to UFHs. Other advantages of LMWH include the convenience of its fixed daily dosing regimens (once or twice), the fact that routine monitoring is not required and its track record of safety and efficacy in practice. The renal clearance for LMWH is dose-independent and the half-life is longer than for UFH (17–21 h). Anti-Xa testing is optional in high-risk patients (renal insufficiency, pregnancy, non-compliance) and its interpretation is product specific.
When administered subcutaneously, fondaparinux undergoes rapid and complete absorption and exhibits a half-life of 17–21 h and is excreted primarily unchanged in the urine in patients with normal renal function. Similar to LMWH and UHF, fondaparinux has been proven to be safe and effective for treatment of DVT and pulmonary embolism (PE). When studied in clinical trials, fondaparinux demonstrates superior efficacy in reducing VTE in various orthopedic surgeries but its incidence of major bleeding is higher compared with LMWH. Of note, the timing of fondaparinux administration has been shown to be associated with its efficacy and outcomes and the incidence of major bleeding. Fondaparinux currently does not have US Food and Drug Administration (FDA) approval for acute coronary syndromes.
14.3 Direct Thrombin Inhibitors
The direct thrombin inhibitors (DTIs) lepirudin and hirudin are naturally occurring anticoagulants derived from the salivary glands of the medicinal leech and have demonstrated efficacy in models of thrombosis. The synthetic analogs of hirudin, bivalirudin, and desirudin exert their anticoagulant activity by direct, selective, and reversible binding to the active catalytic site and the anion-binding site of thrombin, which antagonizes the recognition and binding of fibrinogen at the anion binding exosite 1 of thrombin. Argatroban, which is derived from the amino acid arginine, is another smaller synthetic thrombin inhibitor, which also binds reversibly but non-covalently to an active thrombin site. The currently available DTIs vary significantly in regards to their pharmacologic profiles. Bivalirudin possesses a predictable, dose-dependent anticoagulant activity, has the shortest half-life, and therefore is the most suitable agent in the perioperative space. Patient-specific factors (age, cardiovascular function, and hepatic or renal dysfunction) dictate the choice of DTIs. Comorbidities and organ dysfunction in critically ill patients typically dictate lower infusion rates. Standard monitoring of DTI administration follows the aPTT, with a goal of 1.5–2.5 times control (bivalirudin) and 1.5–3 times control (argatroban) while desirudin does not require routine monitoring. As an alternative to aPTT levels and because of inconsistencies in aPTT measurements, the plasma-diluted thrombin time can be considered.
14.4 Oral Antiplatelet Treatments
Platelets are essential to hemostasis and to the development of a pathological thrombus. In a multifactorial process, initial disruption of the endothelium exposes platelets to the adhesive proteins of the subendothelial matrix. Interactions between the matrix proteins and platelet-receptor glycoproteins enable platelet adhesion and subsequent activation of intracellular signaling pathways that trigger the release of activators such as ADP, serotonin, adrenaline, thrombin, and thromboxane A2. Via G-protein-coupled receptors, these agonists potentiate each other’s actions. Ultimately, the glycoprotein IIb/IIIa complexes on platelets bind to fibrinogen, a process that results in platelet aggregation and can culminate in thrombus formation (◘ Fig. 14.2). In summary, there are multiple steps involved in the adhesion, activation, and aggregation of platelets and more recently developed antiplatelet drugs either target platelet activating factors or associated receptors.
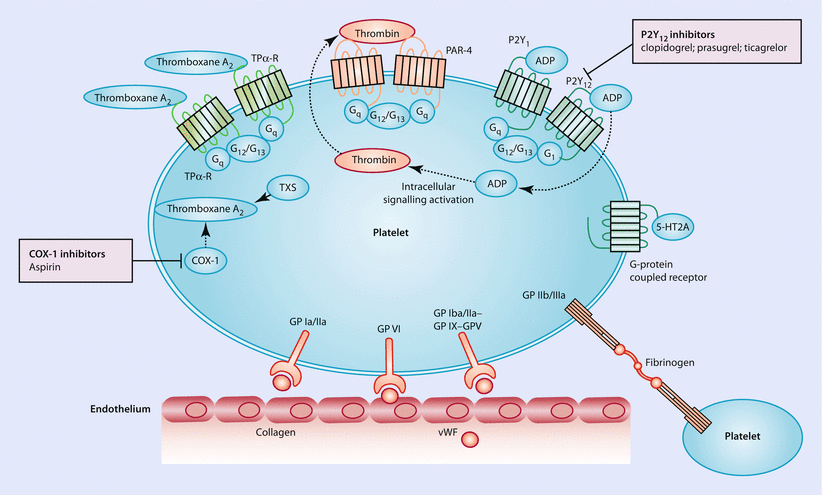
Fig. 14.2
Platelet pathways and commonly used oral antiplatelet treatments such as COX-1 inhibitors, P2Y12 inhibitors and others. Following the disruption of the endothelium, adhesive proteins of the subendothelial matrix (collagen and von Willebrand factor [vWF]) are being exposed. These proteins then interact with platelet-receptor glycoproteins (GP). Intracellular signaling activation results in the release of platelet activators such as ADP, adrenaline, serotonin, thrombin, and thromboxane A2. These agonists bind to G-protein-coupled receptors and further potentiate the process. Ultimately, GP IIb/IIIa binds to fibrinogen and results in platelet aggregation. Abbreviations: 5-HT2A serotonin receptor 2A, COX-1 cyclooxygenase 1, PAR protease-activated receptor, TP-R thromboxane prostanoid receptor, TXS thromboxane A2 synthase, G G-protein. Dotted arrows show movement of molecules (Adapted with permission from Franchi and Angiolillo [2])
14.4.1 Aspirin
Aspirin (acetylsalicylic acid) is the most commonly prescribed antiplatelet drug for prevention of cardiovascular disorders. While low doses of aspirin selectively inhibit cyclooxygenase (COX)-1, resulting in its antiplatelet effects, higher doses of aspirin inhibit both COX-1 and COX-2 leading to anti-inflammatory and analgesic effects.
Aspirin prevents the access of arachidonic acid to its receptor and inhibits production of thromboxane A2 by acetylating a serine residue near the narrow catalytic site of the COX-1 channel.
Repeated low doses of aspirin result in permanent and irreversible thromboxane A2 inhibition throughout the 7–10 day lifetime of anucleated platelets. This is why aspirin can be administered once daily and still remains effective despite its very short half-life (15–20 min). Of note, aspirin also exerts other relevant (indirect) properties such as the reduction of inflammatory cytokines, oxygen radicals, and growth factors relevant.
Absorption of aspirin is rapid and mediated by passive diffusion through gastrointestinal membranes. Single or repeated oral administration of aspirin results in systemic bioavailability of about 45–50%, but the enteric-coated formulation has a much lower bioavailability. With regular aspirin, peak plasma concentrations of aspirin are reached within 30 min after ingestion but up to 4 h after ingestion of the enteric-coated formulation. In patients with acute coronary syndrome (ACS), rapid inhibition of thromboxane A2 can be achieved with a recommended dose of 150–325 mg of aspirin. There appears to be no evidence that patients with ACS benefit from higher versus lower doses of oral aspirin, but lower doses (81–100 mg) have been shown to result in relatively less bleeding than higher doses of aspirin. Alternatively, lower doses of aspirin can also be given intravenously.
The return of platelet function after aspirin administration is related to the natural turnover of platelets. Assuming a daily generation of 10–12% new platelets from megakaryocytes, near normal hemostasis can be seen within 2–3 days after the last aspirin dose in patients with a typical rate of platelet turnover. Variability in inhibition of platelet thromboxane A2, often termed as “aspirin resistance” can be seen in proinflammatory settings (such as in patients with ACS or diabetes) that promote a faster rate of platelet turnover and platelet hyper-reactivity.
14.4.2 P2Y12 Receptor Antagonists
Dual antiplatelet therapy with aspirin and a P2Y12 receptor antagonist, such as clopidogrel, is considered as standard treatment of patients with ACS and those undergoing coronary interventions with placement of intracoronary stents. Although the first-generation P2Y12 receptor antagonist ticlopidine proved to reduce the risk of stent thrombosis compared with standard treatments it was soon replaced with clopidogrel, which had fewer hematological side effects. Common to all PSY12 receptor antagonists is that they either irreversibly (clopidogrel and prasugrel) or reversibly (ticagrelor) block the binding of adenosine diphosphate (ADP) to a specific platelet receptor P2Y12, which consequently inhibits the activation of the glycoprotein (GP) IIb/IIIa complex and platelet aggregation. Improved understanding of the P2Y12 receptor and the pharmacology of antiplatelet drugs resulted in the production of prasugrel and ticagrelor. Compared to clopidogrel, both prasugrel and ticagrelor have faster onset of action and are associated with reduced interpatient variability in platelet inhibition (see ◘ Table 14.2). Although the reasons for the significant interpatient variability associated with clopidogrel are likely multifactorial, they include drug, environmental, and genetic interactions, in addition to clinical factors such as diabetes and obesity.
Table 14.2




Properties of oral P2Y12 inhibitors
Related posts:




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
