Pharmacology
Athena F. Zuppa
KEY POINTS
Scientific Foundations
 An understanding of pharmacokinetics and pharmacodynamics can allow for a rational approach toward prescribing medications for critically ill children.
An understanding of pharmacokinetics and pharmacodynamics can allow for a rational approach toward prescribing medications for critically ill children. Drug absorption, distribution, metabolism, elimination, and the response to medications are impacted by age and disease state.
Drug absorption, distribution, metabolism, elimination, and the response to medications are impacted by age and disease state.Application to Pediatric Intensive Care
 Many medications are prescribed for children based on dosing guidance from adult studies.
Many medications are prescribed for children based on dosing guidance from adult studies. Care providers must be cautious of the high risk of drug interactions and adverse reactions in the intensive care setting.
Care providers must be cautious of the high risk of drug interactions and adverse reactions in the intensive care setting.The word pharmacology is derived from the Greek “pharmacon,” or drug, and “logos,” or science. It is more formally defined as the study of how chemical substances interact with living systems. If these substances have medicinal properties, they are typically referred to as pharmaceuticals. Pharmacology, then, encompasses drug composition, drug properties, interactions, toxicology, and desirable effects that can be used in therapy of diseases. Underlying the discipline of pharmacology are the fields of pharmacokinetics (PK) and pharmacodynamics (PD), which focus on the movement of the various molecular entities contained within a pharmaceutical as it traverses bodily space and the actions of the active moieties once they arrive at the intended physiologic site within the body. Each of these disciplines can be further defined by the underlying processes that dictate specific pathways (e.g., absorption, distribution, metabolism, elimination). Hence, pharmacology is essential to our understanding of how drugs work and how to guide their administration.
Pediatric pharmacotherapy can be challenging due to developmental changes that may alter drug kinetics, pathophysiologic differences that may alter pharmacodynamics, disease etiologies that may differ from those of adults, and other factors that may result in great variation in safety and efficacy outcomes. The situation becomes more convoluted when one considers critically ill children and the paucity of well-controlled pediatric clinical trials in this vulnerable population.  Caregivers who prescribe medications to critically ill children must have some understanding of the basic processes that govern the current dosing recommendations for their patients. In many situations, understanding the source and nature of the data used to support such guidance is helpful from the standpoint of managing expectations of patient response to pharmacotherapy and considering whether dosing modifications should be made. Our comfort in using drugs in critically ill children is often based on the populations previously studied (adults, healthy pediatric patients, etc.) and the knowledge gained from previous experience.
Caregivers who prescribe medications to critically ill children must have some understanding of the basic processes that govern the current dosing recommendations for their patients. In many situations, understanding the source and nature of the data used to support such guidance is helpful from the standpoint of managing expectations of patient response to pharmacotherapy and considering whether dosing modifications should be made. Our comfort in using drugs in critically ill children is often based on the populations previously studied (adults, healthy pediatric patients, etc.) and the knowledge gained from previous experience.
Caregivers who prescribe medications to critically ill children must have some understanding of the basic processes that govern the current dosing recommendations for their patients. In many situations, understanding the source and nature of the data used to support such guidance is helpful from the standpoint of managing expectations of patient response to pharmacotherapy and considering whether dosing modifications should be made. Our comfort in using drugs in critically ill children is often based on the populations previously studied (adults, healthy pediatric patients, etc.) and the knowledge gained from previous experience.A review of the pharmacologic principles that generally guide pharmacotherapy is provided in this chapter, with a focus on specific topics relevant to the research and development of pharmaceuticals in critically ill children. In addition, the current knowledge on dosing medications commonly prescribed for critically ill children is reviewed.
SCIENTIFIC FOUNDATIONS
Pharmacokinetics
Absorption
Absorption is the process of drug transfer from its site of administration to the bloodstream. The rate and efficiency of absorption depend on the route of administration (including but not limited to oral, inhaled, topical). For intravenous (IV) administration, absorption is complete; the total dose reaches the systemic circulation. Drugs administered enterally may be absorbed by either passive diffusion or active transport.
Bioavailability
The bioavailability (F) of a drug is defined by the fraction of the administered dose that reaches the systemic circulation. If a drug is administered intravenously, the bioavailability is 100% and F = 1.0. When drugs are administered by routes other than IV, the bioavailability is usually <100%. Bioavailability is reduced by incomplete absorption, first-pass metabolism, and distribution into other tissues.
Distribution
The volume of distribution (Vd) is a hypothetical volume of fluid through which a drug is dispersed. A drug rarely disperses solely into the water compartments of the body. Instead, most drugs disperse to several compartments (including adipose tissue) and bind to plasma proteins. The total volume into which a drug disperses is called the apparent volume of distribution. This volume is not a physiologic space, but rather a conceptual parameter. It relates the total amount of drug (Drug) in the body to the concentration of drug (Cp) in the blood or plasma:
Figure 22.1 represents the fate of a hypothetical drug after IV administration. After administration, a maximal plasma concentration is achieved, and the drug is immediately distributed. The plasma concentration then decreases over time. This initial phase is called the α-phase of drug distribution, in
which the decline in plasma concentration reflects the distribution of the drug. Once a drug is distributed, it undergoes metabolism and elimination. The second phase is called the β-phase, in which the decline in plasma concentration reflects drug metabolism and clearance. In equations, the terms A and B are intercepts with the Y-axis. The extrapolation of the β-phase defines B. The dotted line is generated by subtracting the extrapolated line from the original concentration line. This second line defines α and A. The serum concentration (C) can be determined using the formula:
which the decline in plasma concentration reflects the distribution of the drug. Once a drug is distributed, it undergoes metabolism and elimination. The second phase is called the β-phase, in which the decline in plasma concentration reflects drug metabolism and clearance. In equations, the terms A and B are intercepts with the Y-axis. The extrapolation of the β-phase defines B. The dotted line is generated by subtracting the extrapolated line from the original concentration line. This second line defines α and A. The serum concentration (C) can be determined using the formula:
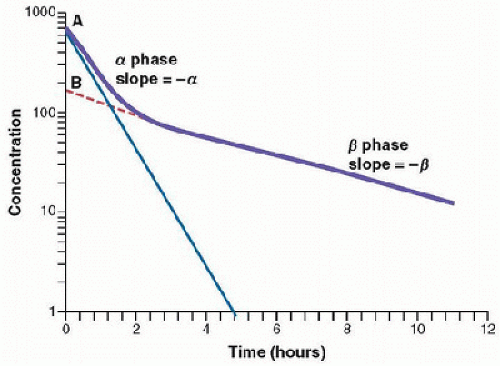 FIGURE 22.1. Semilogarithmic plot of concentration versus time after an IV administration of a drug that follows two-compartment pharmacokinetics. |
The distribution and elimination half-lives can be determined by Equations 22.3 and 22.4, respectively (1,2):
and
For drugs in which distribution is homogenous along the varied physiologic spaces, the distinction between the α and β phases may be subtle and, essentially, a single phase best describes the decline in drug concentration.
Metabolism
The metabolic transformation of drugs is catalyzed by enzymes, and most reactions follow Michaelis-Menten kinetics:
where
V is the rate of drug metabolism
C is the drug concentration
Km is the Michaelis-Menten constant.
In most situations, the drug concentration is much less than Km, and Equation 22.5 simplifies to
In this case, the rate of drug metabolism is directly proportional to the concentration of free drug, and follows first-order kinetics. A constant percentage of the drug is metabolized over time, and the rate of elimination is proportional to the amount of drug in the body. Most drugs used in the clinical setting are eliminated in this manner. The concentration-time curve of drug that follows first-order elimination is represented in Figure 22.2. A semilogarithmic plot results in a straight line (Fig. 22.3).
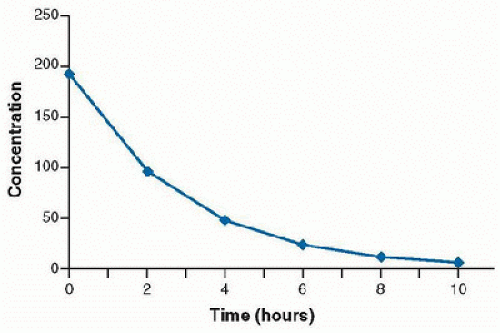 FIGURE 22.2. Concentration versus time profile of a drug demonstrating first-order elimination. |
A few drugs, such as aspirin, ethanol, and phenytoin, are used at higher doses in certain clinical scenarios, resulting in higher plasma concentrations than would be seen at standard doses. In these situations, C is much greater than Km, and Equation 22.5 reduces to:
The enzyme system becomes saturated by a high free-drug concentration, and the rate of metabolism is constant over time. This condition is called zero-order kinetics, and a constant amount of drug is metabolized per unit of time. A large increase in serum concentration can result from a small increase in dose for drugs that follow zero-order elimination. A plot of concentration versus time will result in a straight line (Fig. 22.4). A semilogarithmic plot of concentration versus time demonstrates a convex line (Fig. 22.5).
Phase I and Phase II Biotransformation. The liver is the principal organ of drug metabolism. Other tissues that display considerable drug metabolic activity include the gastrointestinal tract, lungs, skin, and kidneys. Following oral administration, many drugs are absorbed intact from the small intestine and transported via the portal system to the liver, where they are metabolized. This process is called first-pass metabolism and may greatly limit the bioavailability of orally administered drugs.
In general, the metabolic reactions involved in drug metabolism can be classified as either phase I or phase II biotransformation reactions. Biotransformation is the process in which a modification is made on a chemical by an organism. Phase I reactions usually convert a parent drug to a polar
metabolite by introducing or unmasking a more polar site (e.g., -OH, -NH2). Phase I metabolites are generally less active, and because they are more polar, they are more readily excreted. However, many phase I metabolites undergo a subsequent, phase II, reaction in which endogenous substances, such as glucuronic acid, sulfuric acid, or an amino acid, combine with the metabolite to form a highly polar conjugate (conjugation reaction). Many drugs undergo these sequential reactions. However, phase II reactions may precede phase I reactions, as in the case of isoniazid.
metabolite by introducing or unmasking a more polar site (e.g., -OH, -NH2). Phase I metabolites are generally less active, and because they are more polar, they are more readily excreted. However, many phase I metabolites undergo a subsequent, phase II, reaction in which endogenous substances, such as glucuronic acid, sulfuric acid, or an amino acid, combine with the metabolite to form a highly polar conjugate (conjugation reaction). Many drugs undergo these sequential reactions. However, phase II reactions may precede phase I reactions, as in the case of isoniazid.
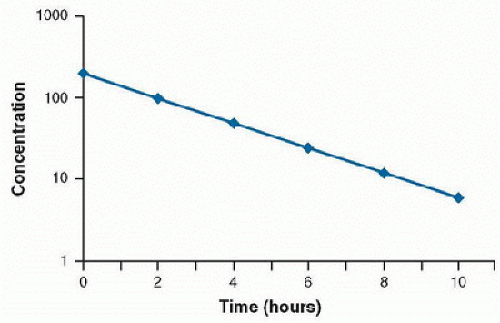 FIGURE 22.3. Semilogarithmic plot of concentration versus time of a drug demonstrating first-order elimination. |
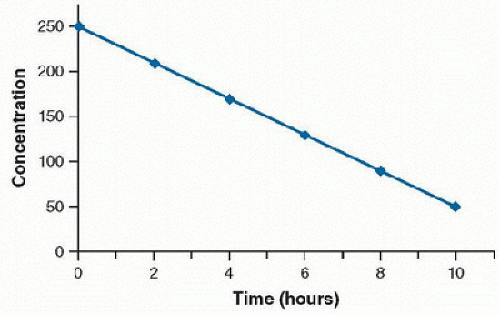 FIGURE 22.4. Concentration versus time of a drug demonstrating zero-order elimination. |
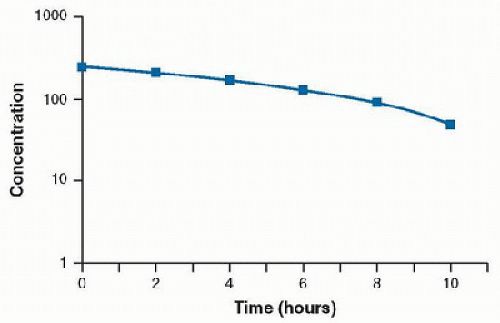 FIGURE 22.5. Semilogarithmic plot of concentration versus time of a drug demonstrating zero-order elimination. |
Phase I reactions are usually catalyzed by enzymes of the cytochrome P450 system. These drug-metabolizing enzymes are located in the lipophilic membranes of the endoplasmic reticulum of the liver and other tissues. Three families, CYP1, CYP2, and CYP3, are responsible for most drug biotransformations. The CYP3A subfamily accounts for >50% of phase I drug metabolism, predominantly by the CYP3A4 subtype. CYP3A4 is responsible for the metabolism of drugs commonly used in the intensive care setting, including acetaminophen, cyclosporine, diazepam, methadone, midazolam, spironolactone, and tacrolimus. Most other drug biotransformations are performed by CYP2D6 (e.g., clozapine, codeine, flecainide, haloperidol, oxycodone), CYP2C9 (e.g., phenytoin, S-warfarin), CYP2C19 (e.g., diazepam, omeprazole, propranolol), CYP2E1 (e.g., acetaminophen, enflurane, halothane), and CYP1A2 (e.g., acetaminophen, caffeine, theophylline, warfarin). Drug biotransformation reactions may be enhanced or impaired by multiple factors, including age, enzyme induction or inhibition, pharmacogenetics, and the effects of other disease states (3,4).
Elimination
Elimination is the process by which a drug is removed or “cleared” from the body. Clearance (CL) is usually considered to be the amount of blood from which all drug is removed per unit of time (volume/time). The kidneys and liver are the main organs responsible for drug clearance. The total body clearance of a drug is equal to the sum of the clearances from all mechanisms, typically partitioned into renal and nonrenal clearance. Most elimination by the kidneys is accomplished through glomerular filtration. Glomerular integrity, the size and charge of the drug, water solubility, and the extent of protein binding determine the amount of drug filtered at the glomerulus. Highly protein-bound drugs are not readily filtered. Glomerular filtration rate has traditionally served as an approximation of renal function.
In addition to glomerular filtration, drugs may be eliminated from the kidneys via active secretion. Secretion occurs predominantly at the proximal tubule, where active transport systems secrete primarily organic acids and bases. Organic acids include most cephalosporins, loop diuretics, methotrexate, nonsteroidal anti-inflammatories, penicillins, and thiazide diuretics. Organic bases include ranitidine and morphine.
As drugs move toward the distal convoluting tubule, the concentration increases. High urine flow rates decrease the concentration of drug in the distal tubule, decreasing the likelihood that a drug will diffuse from the lumen. For both weak acids and bases, the nonionized form of the drug is reabsorbed more readily. Altering the pH (ion trapping) can minimize reabsorption by placing a charge on the drug and preventing its diffusion. For example, salicylate is a weak acid. In case of salicylate toxicity, urine alkalinization places a charge on the molecule and increases its elimination by reducing tubular reabsorption.
The liver also contributes to elimination through metabolism or excretion into the bile. After a drug is secreted in the bile, it may be either excreted into the feces or reabsorbed via enterohepatic recirculation (1,3,4).
The half-life of elimination is the time it takes to clear half of the drug from plasma. It is directly proportional to the Vd and inversely proportional to CL (1):
Renal Dysfunction
Renal failure can impact drug pharmacokinetics through several mechanisms. The binding of acidic drugs to albumin is reduced because accumulated organic acids compete with albumin-binding sites and because uremia changes the structure of albumin. Altered albumin binding leads to altered Vd and, consequently, to altered elimination half-life (4). Renal insufficiency is likely to decrease the clearance of drugs that are normally >30% eliminated unchanged in the urine, which results in a prolonged t1/2β of drugs such as milrinone, digoxin, aminoglycosides, insulin, and others (4).
Hepatic Dysfunction
Drugs that undergo extensive first-pass metabolism may have a significantly higher oral bioavailability in patients with liver failure than in normal subjects. Gut hypomotility may delay the peak response to enterally administered drugs in these patients. Hypoalbuminemia or altered glycoprotein levels may affect the fractional protein binding of acidic or basic drugs, respectively. Altered plasma protein concentrations may affect the extent of tissue distribution of drugs that are normally
highly protein bound. The presence of significant edema and ascites may alter the Vd of highly water-soluble agents, such as aminoglycoside antibiotics. The capacity of the liver to metabolize drugs depends on hepatic blood flow and liver enzyme activity, both of which can be affected by liver disease. In addition, liver disease affects P450 isoforms in a variable manner; some P450 isoforms are more dysfunctional in the context of liver disease than others. This variability in susceptibility leads to the heterogeneous effects of liver disease on P450-dependent drug metabolism (4,5).
highly protein bound. The presence of significant edema and ascites may alter the Vd of highly water-soluble agents, such as aminoglycoside antibiotics. The capacity of the liver to metabolize drugs depends on hepatic blood flow and liver enzyme activity, both of which can be affected by liver disease. In addition, liver disease affects P450 isoforms in a variable manner; some P450 isoforms are more dysfunctional in the context of liver disease than others. This variability in susceptibility leads to the heterogeneous effects of liver disease on P450-dependent drug metabolism (4,5).
Cardiac Dysfunction
Circulatory failure, or shock, can alter the pharmacokinetics of drugs frequently used in the intensive care setting. Drug absorption may be impaired because of bowel wall edema. Passive hepatic congestion may impede first-pass metabolism, resulting in higher plasma concentrations. Peripheral edema inhibits absorption by intramuscular parenteral route. The balance of tissue hypoperfusion versus increased total body water with edema may unpredictably alter Vd. In addition, liver hypoperfusion may alter drug-metabolizing enzyme function, especially flow-dependent drugs such as lidocaine (4).
As children develop and grow, changes in body composition, development of metabolizing enzymes, and maturation of renal and liver function all have an impact on drug disposition (1,2).
Renal
Glomerular filtration and tubular secretion are significantly reduced in the premature and full-term neonate, as compared with older children. Maturation of renal function is a dynamic process that begins during fetal life and is complete by early childhood. Maturation of tubular function is slower than that of glomerular filtration. The glomerular filtration rate is ~2-4 mL/min/1.73 m2 in term neonates, but it may be as low as 0.6-0.8 mL/min/1.73 m2 in preterm neonates. The glomerular filtration rate increases rapidly during the first 2 weeks of life and continues to rise until adult values are reached at 8-12 months of age. For drugs that are dependent on renal elimination, impaired renal function decreases clearance and increases the half-life. Therefore, for drugs that are primarily eliminated by the kidney, dosing should be performed in an age-appropriate fashion that takes into account maturational changes in kidney function (1,2,6).
Hepatic
Hepatic biotransformation reactions are substantially reduced in the neonatal period. At birth, the cytochrome P450 system is 28% that of the adult (1,2). The expression of phase I enzymes (such as the P450 cytochromes) changes markedly during development. CYP3A7, the predominant CYP isoform expressed in fetal liver, peaks shortly after birth and then declines rapidly to levels that are undetectable in most adults. Within hours after birth, CYP2E1 activity surges, and CYP2D6 becomes detectable soon thereafter. CYP3A4 and CYP2C (CYP2C9 and CYP2C19) appear during the first week of life, whereas CYP1A2 is the last hepatic CYP to appear, at 1-3 months of life (6). The ontogeny of phase II enzymes is less well established than the ontogeny of reactions that involve phase I enzymes. Available data indicate that the individual isoforms of uridine diphosphate glucuronosyltransferase (UGT) have unique maturational profiles with pharmacokinetic consequences. For example, the glucuronidation of acetaminophen (a substrate for UGT1A6 and, to a lesser extent, UGT1A9) is decreased in newborns and young children, as compared with adolescents and adults. Glucuronidation of morphine (a UGT2B7 substrate) can be detected in premature infants as young as 24 weeks of gestational age (6).
Gastrointestinal
Overall, the rate at which most drugs are absorbed from the gastrointestinal tract is slower in neonates and young infants than in older children. As a result, the time required to achieve maximal plasma levels of enterally administered drugs is longer in the very young (6). The effect of age on enteral absorption is not uniform, and it is difficult to predict (1,2). Gastric emptying and intestinal motility are the primary determinants of the rate at which drugs are presented to, and dispersed along, the mucosal surface of the small intestine. At birth, the coordination of antral contractions improves, resulting in a marked increase in gastric emptying during the first week of life. Similarly, intestinal motor activity matures throughout early infancy, with consequent increases in the frequency, amplitude, and duration of propagating contractions (6).
Changes in the intraluminal pH in different segments of the gastrointestinal tract can directly affect both the stability and the degree of ionization of a drug, thus influencing the relative amount of drug available for absorption. During the neonatal period, intragastric pH is relatively elevated (>4). Thus, oral administration of acid-labile compounds such as penicillin G produces greater bioavailability in neonates than in older infants and children (7). In contrast, drugs that are weak acids, such as phenobarbital, may require larger oral doses in the very young to achieve therapeutic plasma levels. Other factors that impact the rate of absorption include age-associated development of villi, splanchnic blood flow, changes in intestinal microflora, and intestinal surface area (6).
Body Composition
Age-dependent changes in body composition alter the physiologic spaces into which a drug may be distributed (6). The percentage of total body water drops from ~85% in premature infants to 75% in full-term infants to 60% in the adult. Extracellular water decreases from 45% in the infant to 25% in the adult. Total body fat in the premature infant can be as low as 1%, as compared to 15% in the normal term infant. Many drugs are less bound to plasma proteins in the neonate and infant than in the older child (1,2).
Much of drug distribution is a result of simple passive diffusion along concentration gradients and subsequent binding of the drug to tissue components. However, tissue transporters capable of producing a biologic barrier also contribute to drug distribution. An example is P-glycoprotein, a member of the ATP-binding cassette family of transporters that functions as an efflux transporter capable of extruding selected substances from cells. The expression and localization of P-glycoprotein in specific tissues facilitates its ability to limit the cellular uptake of selected substrates to these sites (e.g., the blood-brain barrier, hepatocytes, renal tubular cells, and enterocytes). Limited data are available regarding the ontogeny of P-glycoprotein expression in humans. A single study of the expression of P-glycoprotein in the central nervous system (CNS) using tissue obtained postmortem from neonates born at 23-42 weeks of gestational age suggests a pattern of localization similar to that in adult mice late in gestation and at term. However, the level of P-glycoprotein expression appeared to be lower than that in adults (8). Limited data in neonates suggest that the passive diffusion of drugs into the CNS is age dependent, as reflected by the progressive
increase in the ratios of brain phenobarbital to plasma phenobarbital from 28 to 39 weeks of gestational age, demonstrating the increased transport of phenobarbital into the brain (9).
increase in the ratios of brain phenobarbital to plasma phenobarbital from 28 to 39 weeks of gestational age, demonstrating the increased transport of phenobarbital into the brain (9).
Pharmacodynamics
Pharmacodynamics, in general terms, seeks to define what the drug does to the body (i.e., the effects or response to drug therapy). Pharmacodynamics modeling attempts to characterize measured, physiologic parameters before and after drug administration, with the effect defined as the change in a physiologic parameter relative to its predose or baseline value. Baseline is defined as the physiologic parameter without drug administration and may be complicated in certain situations due to diurnal variations. Efficacy can be defined numerically as the expected sum of all beneficial effects following treatment. Similarly, toxicity can be characterized by either the time course of a specific toxic event or the composite of detrimental responses attributed to a common toxicity.
Overview
Pharmacodynamic response to drug therapy evolves only after active drug molecules reach their intended site(s) of action. Hence, the link between pharmacokinetic and pharmacodynamic processes is implicit. Differences in pharmacodynamic time course among drug entities can be broadly associated with the nature of the concentration-effect relationship as being direct (effect is directly proportional to concentration at the site of measurement, usually the plasma) or indirect (effect exhibits some type of temporal delay with respect to drug concentration either because of differences between the drug’s site of action and measurement of effect or because the effect of interest results after other physiologic or pharmacologic conditions are satisfied).
Direct effect relationships are easily observed with cardiovascular agents. Pharmacologic effects such as blood pressure, ACE (angiotensin-converting-enzyme) inhibition, and inhibition of platelet aggregation can be characterized by direct response relationships. Such relationships can usually be defined by three typical patterns: linear, hyperbolic maximum effect observed (Emax), and sigmoid Emax functions (Fig. 22.6). In each case, the plasma concentration and drug concentration at the effect site are proportional. Likewise, the concentration-effect relationship is assumed to be independent of time.
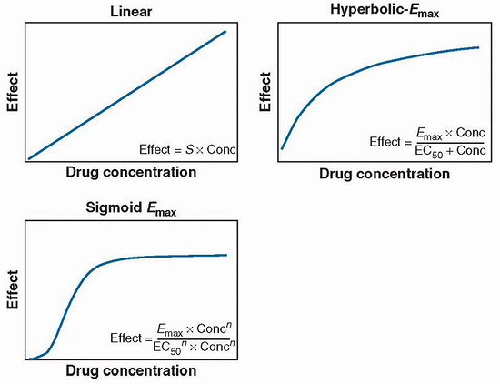 FIGURE 22.6. Representative pharmacodynamic relationships for drugs that exhibit direct responses: linear, hyperbolic, and sigmoid Emax relationships shown. S is the slope of the linear response; Emax refers to the maximum effect observed; EC50 refers to the concentration at which 50% of the maximal response is achieved, and n is the degree of sigmoidicity or shape factor (sometimes referred to as the Hill coefficient). |
Other drugs exhibit an indirect relationship between concentration and response. In this case, the concentration-effect relationship is time dependent. One explanation for such effects is hysteresis. Hysteresis refers to the phenomenon in which a time lapse exists between the cause and its effect. With respect to pharmacodynamics, this time lapse most often indicates a situation in which a delay exists in equilibrium between plasma drug concentration and the concentration of active substance at the effect site. Three broad conditions can account for this phenomenon: the active drug response site is not in the central compartment, the mechanism of drug action involves new protein synthesis, or the particular drug has active metabolites.
More complicated models (indirect response models) have been used to express the same observations but typically necessitate a greater understanding of the underlying physiologic process (e.g., cell trafficking, enzyme recruitment, etc.). The salient point is that pharmacodynamic characterization and dosing guidance derived from such characterization tend to be more informative than drug concentrations alone.
Pharmacogenomics
Pharmacogenomics is the study of how an individual’s genetic inheritance affects his or her response to drugs. Pharmacogenomics holds the promise that drugs might one day be tailored to individuals and adapted to each person’s own genetic makeup. Environment, diet, age, lifestyle, and state of health all can influence a person’s response to medicines, but understanding an individual’s genetic composition is thought to be a key to creating personalized drugs with greater efficacy and safety. Pharmacogenomics combines traditional pharmaceutical sciences, such as biochemistry, with annotated knowledge of genes, proteins, and single nucleotide polymorphisms (see Chapter 18).
Benzodiazepines
Benzodiazepines are often used to provide sedation and amnesia. Benzodiazepines exert their anxiolytic, amnestic, anticonvulsant, and muscle-relaxing effects through interaction at specific binding sites on neuronal γ-aminobutyric acid (GABA) receptors (10). Benzodiazepines facilitate the inhibitory action of GABA on neuronal impulse transmission. The potency of individual medications is determined by their receptor affinity. Benzodiazepines enhance inhibitory synaptic transmission by mimicking and increasing the actions of GABA-mediated chloride influx at the GABA receptor.
Chronic administration of benzodiazepines can lead to decreased receptor activity and drug tolerance. Tolerance is a common finding in ICU patients who receive benzodiazepines or other sedative agents for periods >24 hours. Withdrawal syndromes have been reported with the cessation of midazolam and other benzodiazepine infusions. Risk factors for acute withdrawal include high infusion rates, prolonged duration, and abrupt cessation. For these reasons, gradual tapering of sedative infusions and substitution with longer-acting agents (e.g., diazepam) are suggested to reduce the chance of withdrawal reactions. Benzodiazepines also are noted for occasionally producing paradoxical reactions, including increased agitation and delirium (11).
Diazepam
Diazepam is highly lipid soluble and protein bound, and distributes quickly into the brain. It is available in IV and oral preparations. Diazepam administration results in antegrade but not retrograde amnesia. It reduces the cerebral metabolic rate for oxygen consumption and thus decreases cerebral blood flow in a dose-dependent manner. Diazepam, like the other benzodiazepines, raises the seizure threshold (11). After enteral dosing, diazepam demonstrates a bioavailability of 100%. It has a Vd of 1.1-2.9 L/kg (11,12) and is 98% protein bound (12). Diazepam is metabolized by hepatic microsomal enzymes (CYP2C19) to active compounds such as desmethyldiazepam and oxazepam. Desmethyldiazepam has a long elimination half-life of 100-200 hours and is eliminated by the kidneys. Oxazepam has an elimination half-life of 10 hours. The elimination half-life of diazepam averages 72 hours, varies widely, and is increased in the elderly, neonates, and patients with liver disease. Metabolism is also affected by genetics, gender, endocrine status, nutritional status, smoking, and concurrent drug therapy (11). The mean plasma clearance is 0.27-0.37 mL/kg/min and is independent of liver blood flow (13).
Diazepam alone has minimal cardiovascular depressant effects, although systemic vascular resistance is reduced slightly, producing a small decline in arterial blood pressure. Respiratory drive is minimally decreased by diazepam alone but is profoundly depressed when diazepam (or another benzodiazepine) is used in combination with opioids. Diazepam elimination is decreased by such drugs as cimetidine, fluconazole, and valproic acid (11).
Midazolam
Midazolam is three to four times more potent than diazepam (11). It is rapidly absorbed after administration of the oral syrup formulation, with adolescents absorbing the drug at approximately half the rate observed in younger children (ages 2 to <12 years). In children 6 months to 16 years, the absolute bioavailability of oral midazolam averaged 36%, with a very broad range (9%-71%). No relationship between midazolam bioavailability and age was observed (14). The pKa of midazolam (6.1) is especially important because it permits a conformational change in midazolam’s structure, depending on pH. As currently marketed, midazolam is buffered to a pH of 3.5, opening its imidazole ring and increasing its water solubility. At physiologic pH, however, the diazepine ring closes rapidly and midazolam becomes lipid soluble. As a consequence, the effects of midazolam are rapid.
Midazolam is 95% protein bound, with a Vd of 1.9 L/kg (11). Midazolam undergoes extensive metabolism by the cytochrome P4503A subfamily (e.g., CYP3A4 and CYP3A5) to major (1-OH-midazolam) and minor (4-OH-midazolam) hydroxylated metabolites. 1-OH-midazolam is equipotent to midazolam. Both hydroxylated metabolites are subsequently glucuronidated by uridine diphosphate glucuronosyltransferases (UGTs). 1-OH-midazolam-glucuronide also appears to have sedative properties when concentrations are high, as has been observed in adult patients with renal failure (15).
In healthy children 6 months to <2 years, 2 years to <12 years, and 12 years to <16 years, midazolam clearance was 11.3, 10, and 9.3 mL/kg/min, respectively (14). Adult clearance is estimated at 6.6 mL/kg/min (12). CYP3A4/5 activity reaches adult levels at between 3 and 12 months of postnatal age (16). Developmental differences in CYP3A activity may therefore alter the pharmacokinetics of midazolam in pediatric intensive care patients of different ages. Similarly, the UGTs exhibit developmental changes in activity. However, because the specific UGTs involved in the conjugation of 1-OH-midazolam are not yet known, the impact of ontogeny on this reaction remains to be determined (15).
Less than 1% of midazolam is excreted unchanged in the urine. The elimination half-life of midazolam is 2 hours in young, healthy adults but increases rapidly in the elderly and following major surgery (11). 1-OH-midazolam-glucuronide is renally excreted, with an elimination half-life of 1 hour with normal renal function (15). This results in high concentrations in the presence of renal failure and is responsible for prolonged sedation (13).
Midazolam is metabolized by hepatic microsomal oxidation. The oxidative pathway is susceptible to many factors, including hepatic disease, advanced age, and drug inhibition. The most dramatic changes in the pharmacokinetics of midazolam in the critically ill may result from altered hepatic metabolism. Accumulation occurs in critically ill patients at the peak of their illness, with low or absent concentrations of 1-hydroxymidazolam, suggesting failure of liver metabolism (17). A number of drugs, including cimetidine, erythromycin, propofol, and diltiazem, have been reported to delay midazolam metabolism and therefore increase its duration of effect.
The accumulation of the active metabolite also may be important in some ICU patients. As with other benzodiazepines, midazolam causes dose-related respiratory depression and, in large doses, can cause vasodilation and hypotension (11). Even small doses of midazolam, when combined with an opioid, may lead to hypotension in infants in the immediate postoperative period after cardiac surgery.
As with many highly lipid soluble drugs, midazolam accumulates in peripheral tissues and in the bloodstream rather than being metabolized after continuous infusion for extended time periods. When the infusion is discontinued, peripheral tissue stores release midazolam back into the plasma, and the duration of clinical effect can be prolonged. Obese patients with larger volumes of distribution and elderly patients with decreased hepatic and renal function may be at higher risk for prolonged sedation from midazolam (18).
Lorazepam
Lorazepam is the least lipid soluble of the three benzodiazepines and traverses the blood-brain barrier most slowly, resulting in delayed onset and prolonged duration of effect (11). Lorazepam is well absorbed orally and demonstrates a bioavailability of 93% after oral administration (12). It is absorbed rapidly after intramuscular injection, and plasma concentrations peak within 60 minutes. It is ~90% protein bound and has a Vd of 2 L/kg. Lorazepam is metabolized to inactive products by hepatic glucuronidation. The pharmacokinetics of lorazepam does not change significantly in the elderly or in critically ill populations. The elimination half-life ranges from 10 to 20 hours but is prolonged by liver and end-stage kidney disease (11). CL is estimated at 1.1 mL/kg/min (12). Because lorazepam is insoluble in water, it is manufactured with a vehicle that contains polyethylene glycol, benzyl alcohol, and propylene glycol. This drug vehicle may be associated with lactic acidosis, hyperosmolar coma, and a reversible nephrotoxicity after high doses or prolonged infusions and a “gasping syndrome” in neonates (11).
Flumazenil
Flumazenil acts at the benzodiazepine binding site on the GABA receptor to antagonize the effects of benzodiazepine agonists. Flumazenil is chemically and structurally similar to other benzodiazepine receptor agonists. Flumazenil produces a reversal of benzodiazepine-induced sedative and amnestic effects. Clinical effects are seen immediately after IV administration. Flumazenil does not reverse the effects of opioids, barbiturates, alcohol, or other GABA-mimetic agents (11). Flumazenil is short acting, and careful clinical observation is crucial if the therapeutic intent is to avoid the recurrence of benzodiazepine-induced sedation. Repeated administrations may be necessary to maintain its antagonistic action (19).
Flumazenil is 40%-50% plasma protein bound, with a Vd of 0.6-1.6 L/kg (12,19). It is cleared rapidly from the plasma by hepatic metabolism. Less than 0.2% of an IV dose is recovered unchanged in the urine, and three metabolites of flumazenil have been identified (19). The elimination half-life of flumazenil is ~1 hour, which is significantly shorter than that of many of the benzodiazepine compounds used clinically. The CL is estimated at 17 mL/kg/min (12). Flumazenil can precipitate withdrawal and/or seizures in patients with benzodiazepine dependence caused by chronic exposure (11).
Barbiturates
Barbiturates are weak acids that are absorbed and rapidly distributed to all tissues and fluids, with high concentrations in the brain, liver, and kidneys. High lipid solubility is the dominant factor in the distribution of barbiturates within the body. The more lipid soluble the barbiturate, the more rapidly it penetrates all tissues of the body. Barbiturates are bound to plasma and tissue proteins to varying degrees, with the degree of binding increasing directly as a function of lipid solubility. Barbiturates are absorbed in varying degrees following oral, rectal, or parenteral administration.
Pentobarbital
Pentobarbital is a commonly used barbiturate for sedation, hypnosis, and intracranial pressure (ICP) management. Pentobarbital has two enantiomeric forms: R– and S-pentobarbital. The S-enantiomer causes a longer duration of sedation than does the R-enantiomer in humans. High-dose pentobarbital infusions have been advocated as an effective adjunct in controlling persistent intracranial hypertension after severe head trauma in patients who are refractory to conventional therapy. Pentobarbital’s beneficial effects in patients with increased ICP include both a direct vasoconstriction of cerebral blood vessels and a reduction of blood flow through reduction in cerebral metabolic rate. Pentobarbital has a potent effect on GABA-sensitive chloride channels and is a potent CNS depressant. Following IV administration, the onset of action is almost immediate for pentobarbital. Pentobarbital enters the brain more rapidly than do phenobarbital or diazepam and is a very potent antiepileptic drug. Pentobarbital has also been recommended as a sedative agent for diagnostic imaging studies (20,21). Pentobarbital administration has potential respiratory-depressant and hypotensive side effects. Myocardial depression contributes to the hypotension and may require epinephrine infusion in patients with high ICP to maintain cerebral perfusion pressure. Phenylephrine administration alone for pentobarbital-induced hypotension may not overcome the myocardial depression nor improve the cerebral perfusion pressure. Tolerance may develop with prolonged use.
Pentobarbital is metabolized primarily by the hepatic microsomal enzyme system, and the metabolic products are excreted in the urine and, less commonly, in the feces (22). The CL of the S-enantiomer is less than that of the R-enantiomer (43 vs. 32 mL/min), and the Vd of the S-enantiomer is slightly less than the R-enantiomer (1.1 vs. 1.2 L/kg). The S-enantiomer is also more strongly protein bound in plasma (73.5% vs. 63.4% for the R-enantiomer) (23). In a study of seven healthy adult men and women, 100 mg of pentobarbital was administered as an IV bolus. A two-compartment model determined the central Vd to be 0.44 L/kg, with a peripheral Vd of 0.56 L/kg (Vd = 1 L/kg). The elimination half-life was 22.3 hours (24




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
