(25.1)
where concentration is C, t is time after the dose, A is the concentration at time 0 for the distribution rate represented by the broken line graph with the steepest slope, α is the rate constant for distribution, B is the concentration at time 0 for the terminal elimination rate, and β is the rate constant for terminal elimination. Rate constants indicate the rate of change in concentration and correspond to the slope of the line divided by 2.303 (log e 10) for logarithm concentration versus time.
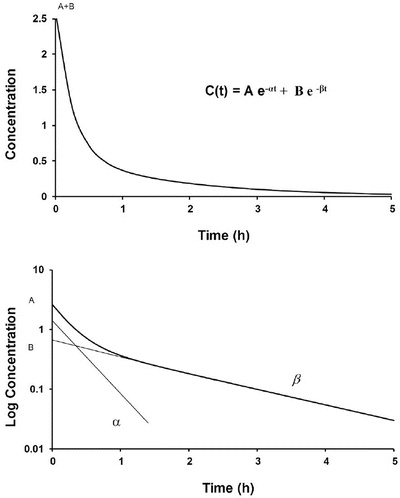
Fig. 25.1
A time–concentration profile of a two-compartment model (upper panel). This profile is shown in a semilogarithmic graph on the lower panel. The initial rapid decrease in serum concentration reflects distribution and elimination followed by a slower decrease due to elimination. Subtraction of the initial decrease in concentration due to elimination using the concentrations from the elimination line extrapolated back to time 0 at B produces the lower line with a steep slope = α (distribution rate constant)/2.303. The terminal elimination phase has a slope = β (elimination rate constant)/2.303
Such two-compartment or biphasic kinetics are frequently observed after IV administration of drugs that rapidly distribute out of the central compartment (V 1) to a peripheral compartment (V 2) [10, 11]. In such situations, the initial rapid decrease in concentration is referred to as the α distribution phase and represents distribution to the peripheral (tissue) compartments in addition to drug elimination. The terminal (β) phase begins after the inflection point in the line when elimination starts to account for most of the change in drug concentration. To determine the initial change in concentration due to distribution, the change in concentration due to elimination must be subtracted from the total change in concentration. The slope of the line representing the difference between these two rates is the rate constant for distribution.
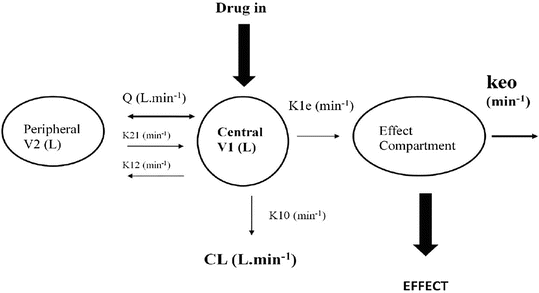
These parameters (A, B, α, β) have little connection with the underlying physiology, and an alternative parameterisation is to use a central volume and three rate constants (k 10, k 12, k 21) that describe drug distribution between compartments. Another common method is to use two volumes (central, V 1; peripheral, V 2) and two clearances (CL, Q). Q is the inter-compartment clearance, and the volume of distribution at steady state (V ss) is the sum of V 1 and V 2 (Fig. 25.2). Computers have made nonlinear regression techniques to directly estimate parameters easier through iterative techniques using least squares curve fitting. Models with two or more compartments are now commonly solved using differential equations rather than graphical techniques, e.g. for a two-compartment mammillary model comprising a central compartment with volume V 1 and concentration C 1 and a peripheral compartment (V 2, C 2) with drug input (ratein)
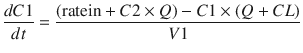
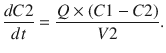
A series of similar differential equations can be written and solved for models with more than two compartments.
Fig. 25.2
A two-compartment model with an additional compartment used to describe concentration in the effect compartment. The effect compartment concentration is not the same as the blood or serum concentration and is not a real measurable concentration. It has negligible volume and contains negligible blood. A single first-order parameter (Keo) describes the equilibration rate between the central and effect compartments (see text for explanation)
(25.2)
(25.3)
Paediatric PK Parameter Sets
Most TCI techniques use propofol and remifentanil as the principle drugs for induction and maintenance of anaesthesia. Popular paediatric programmes used for propofol infusion targeting a plasma concentration are based on data from Marsh et al. [12] and Gepts et al. [13] (Diprifusor) and Kataria et al. [8] or Absalom et al. [14] (Paedfusor). Concentration can be predicted based on the reported parameters. These parameter sets are commonly termed ‘models’ and named after the author who reported them (e.g. ‘Kataria model’). Parameter estimates (e.g. CL, Q, V 1 and V 2) are different for each parameter set (Table 25.1). Covariate influences that contribute variability such as severity of illness are often unaccounted for; the volume in the central compartment, for example, is increased in children after cardiac surgery [15]. Even weight or age, the commonest sources of variability [16], may be omitted from parameter estimates. Both the administration method (intravenous bolus or infusion) [17] and the collection of venous blood for assay rather than arterial blood will have influence on PK parameter estimates in the early phase when movement of drug into the effect compartment is occurring. Time–concentration profiles (Fig. 25.3) and context-sensitive half-lives will differ depending on which parameter set is used [18].
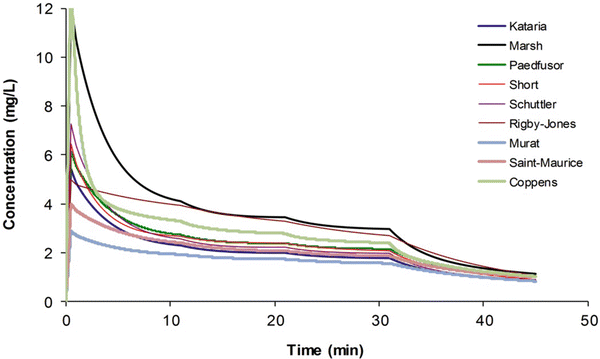
Table 25.1
Propofol parameter estimates for a 20 kg child
Parameter | Kataria [8] | Paedfusor [14] | Short [284] | Schuttler [55] | Rigby-Jones [15] | Murat [285] | Saint-Maurice [286] | Coppens [287] | |
|---|---|---|---|---|---|---|---|---|---|
V 1 (L) | 10.4 | 4.56 | 9.16 | 8.64 | 7.68 | 11.68 | 20.6 | 14.44 | 3.48 |
V 2 (L) | 20.2 | 9.28 | 18.98 | 10.8 | 20.74 | 26.68 | 19.4 | 35.6 | 4.68 |
V 3 (L) | 164 | 58.04 | 116.58 | 69.4 | 264.82 | 223.86 | 121.74 | 168 | 19.02 |
CL1 (L/min) | 0.68 | 0.542 | 0.568 | 0.836 | 0.56 | 0.444 | 0.98 | 0.62 | 0.78 |
CL2 (L/min) | 1.16 | 0.51 | 1.044 | 1.22 | 1.036 | 0.32 | 1.34 | 1.24 | 2.04 |
CL3 (L/min) | 0.52 | 0.192 | 0.384 | 0.34 | 0.46 | 0.268 | 0.4 | 0.22 | 0.66 |
Fig. 25.3
Simulated time–concentration profiles for propofol using differing parameter sets. A 3 mg kg−1 bolus was administered and the infusions were administered as for an adult (10-8-6 regimen) (Published with permission of the publisher. Original source: Anderson BJ. La farmacología de la anestesia total intravenosa en pediatría. Rev Colomb Anestesiol. 2013;41(3):205–214. Copyright © 2013 Sociedad Colombiana de Anestesiología y Reanimación. Publicado por Elsevier España, S.L. Todos los derechos reservados [730])
Validation studies for these differing parameter sets are few. The Paedfusor has been examined [14] and reported to have a MDPE (median performance error, bias) of 4.1 % and a MDAPE (median absolute performance error, precision) of 9.7 % over the age range investigated (1–15 years). A later study suggested that all except Marsh performed acceptably in children 3–26 months [18]. Others have described a poor fit for Kataria, the most widely used model [19]. However, clearance (L h−1 kg−1) decreases with age, and MPE is minimised at low CL and exaggerated at higher values. Evaluating models outside of the age range that they were determined from will increase bias and worsen precision.
Adult remifentanil PK parameters [20] continue to be used in TCI devices for all ages, despite an increasing knowledge about this drug in children [21]. There is an element of safety with this approach because both volume of distribution [22] and clearance (expressed as mL min−1 kg−1) [23] decrease with age from adulthood and because the elimination half-life is small with a constant context-sensitive half-life. The larger volume of distribution results in lower peak concentrations after bolus; the higher clearance in children results in lower plasma concentration when infused at adult rates expressed as mg min−1 kg−1. However, remifentanil PK can be described in all age groups by simple application of an allometric size model (see below) [23]. This standardised clearance of 2790 mL/min/70 kg−1 is similar to that reported by others in children [22, 24] and adults [20, 25]. The smaller the child, the greater the clearance when expressed as mL/min/kg. Owing to these enhanced clearance rates, smaller (younger) children will require higher remifentanil infusion rates than larger (older) children and adults to achieve equivalent blood concentrations.
Half-Life
Half-life, the t ime for a drug concentration to decrease by one half, is a familiar parameter used to describe the kinetics of many drugs that demonstrate exponential decay. Half-life (T 1/2) helps describe this first-order kinetic process, because the same proportion or fraction of the drug is removed during equal periods of time.
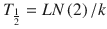
(25.4)
However, half-life is a poor parameter for a drug described using two compartments and may be poorly estimated in drugs with slow absorption after enteral dosing. Half-life does not predict dosing schedule, that is, predicted by effect duration [26]. Half-life is confounded by both clearance and volume; if the two are changing independently with age, the half-life may be the same in neonates as in adults even though clearance is immature in neonates.
A more useful concept for IV drugs used in anaesthesia is that of the context-sensitive half-time (CSHT) where ‘context’ refers to infusion duration. This is the time required for the plasma drug concentration to decline by 50 % after terminating infusion [27]. The CSHT is the same as the elimination half-life for a one-compartment model and does not change with infusion duration.
Context-sensitive half-time may be independent of infusion duration (e.g. remifentanil 2.5 min), moderately affected (propofol 12 min at 1 h, 38 min at 8 h in adults), or display marked prolongation (e.g. fentanyl 1 h at 24 min, 8 h at 280 min). This is due to return of drug to central from peripheral compartments after ceasing infusion. Peripheral compartment sizes and clearances differ in children from adults, and at termination of infusion more or less drug may remain in the body for any given plasma concentration than in adults. The context-sensitive half-time for children given propofol, for example, is longer (Fig. 25.4) [7]. The context-sensitive half-time gives insight into the pharmacokinetics of a drug, but the parameter may not be clinically relevant; the percentage decrease in concentration required for recovery of drug effect is not necessarily 50 % (Fig. 25.5).
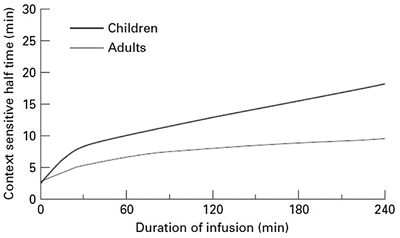
Fig. 25.4
The context-sensitive half-time for children and adults. From McFarlane CS, Anderson BJ, Short TG (Reproduced from: The use of propofol infusions in paediatric anaesthesia: a practical guide. Pediatr Anesth Paediatr Anaesth 1999; 9: 209–216 [7], with kind permission from John Wiley and Sons)
Fig. 25.5
The effect observed when the concentration decreases by 50 % depends on the shape of the concentration–response curve and where the initial concentration sits on that curve. In the example shown decreasing concentration from 40 mg L−1 to 20 mg L−1 will result in no effect change, while decreasing from 20 mg L−1 to 10 mg L−1 will have a minimal change. Impact will certainly be relevant when the concentration changes are around the EC50. This scenario is commonly seen when a large dose of a neuromuscular blocking drug is administered. Dose determines duration of action (Reproduced from Anderson BJ, Holford NH. Tips and traps analyzing pediatric PK data. Pediatr Anesth 2011; 21: 222–37, with kind permission from John Wiley and Sons [26])
Zero-Order Kinetics
The elimination of some drugs occurs with loss of a constant amount per time, rather than a constant fraction per time. Such rates are termed zero order and because e 0 = 1. Zero-order (also known as Michaelis–Menten) kinetics may be designated saturation kinetics, because such processes occur when excess amounts of drug saturate the capacity of metabolic enzymes or transport systems. Ethyl alcohol is a classic example. In this situation, only a constant amount of drug is metabolised or transported per unit of time. If kinetics are zero order, a graph of serum concentration versus time is linear on linear–linear axes and is curved when graphed on linear–logarithmic (i.e. semilogarithmic) axes. Clearance is determined by the maximum rate of metabolism (V max), the Michaelis–Menten constant (K m ) and the concentration (C):
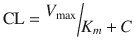
Clinically, first-order elimination may become zero order after administration of excessive doses or prolonged infusions. Certain drugs administered to neonates (with immature clearance pathways) exhibit zero-order kinetics at therapeutic doses and may accumulate to excessive concentrations, including thiopentone, theophylline (Fig. 25.6), caffeine, diazepam, furosemide and phenytoin. Elimination may also be termed ‘mixed order’ (i.e. first order at low concentrations and zero order after enzymes are saturated at higher concentrations). For these drugs, a small increment in dose may cause disproportionately large increments in serum concentrations.
(25.5)
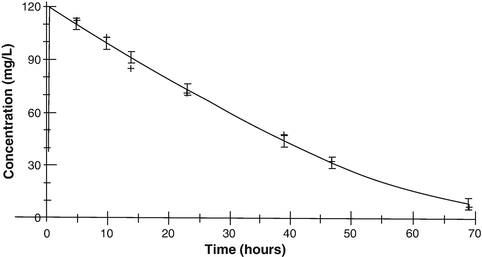
Fig. 25.6
Theophylline elimination in an infant given theophylline 80 mg/kg. Michaelis–Menten kinetics are exemplified by the straight line of the slope of elimination at concentrations above 35 mg/L (Reproduced from Anderson BJ, Holford NH, Woollard GA. Aspects of theophylline clearance in children. Anaesth and Intensive Care 1997; 25: 497–501, with the kind permission of the Australian Society of Anaesthetists [151])
Repetitive Dosing and Drug Accumulation
When multiple doses are administered, the dose is usually repeated before complete elimination of the previous one (Fig. 25.7). In this situation, peak and trough concentrations increase until a steady-state concentration (C ss) is reached. The average C ss can be calculated as follows:
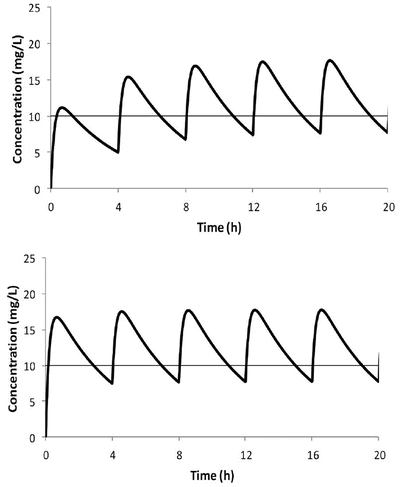
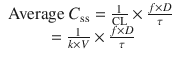
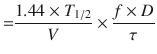
Fig. 25.7
Time–concentration profile for acetaminophen in a child given 250 mg 4 hourly (upper panel). The average C ss of 10 mg/L is reached after three doses. The use of a loading dose rapidly achieves the target concentration (lower panel)
(25.6)
(25.7)
In Eqs. (25.6) and (25.7), f is the fraction of the dose that is absorbed, D is the dose, τ is the dosing interval in the same units of time as the elimination half-life, k is the elimination rate constant, and 1.44 equals 1/Ln(2). The magnitude of the average C ss is directly proportional to a ratio of T 1/2/τ and D.
Steady State
Steady state occur s when the amount of drug removed from the body between doses equals the rate of administration. This can be simplistically described as:
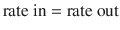
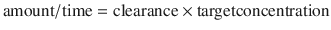
Five half-lives are usually required for drug elimination and distribution among tissue and fluid compartments to reach equilibrium. When all tissues are at equilibrium (i.e. steady state), the peak and trough concentrations are the same after each dose. However, before this time, constant peak and trough concentrations after intermittent doses, or constant concentrations during drug infusions, do not prove that a steady state has been achieved because the drug may still be entering and leaving deep tissue compartments. During continuous infusion, the fraction of steady-state concentration that has been reached can be calculated in terms of multiples of the drug’s half-life. After three half-lives, the concentration is 88 % of that at steady state (Table 25.2).
(25.8)
(25.9)
Table 25.2
Exponential decay and half-life
Half-lives elapsed | Fraction remaining | Percentage remaining | Percentage gone |
|---|---|---|---|
0 | 1 | 100 | 0 |
1 | 1/2 | 50 | 50 |
2 | 1/4 | 25 | 75 |
3 | 1/8 | 12.5 | 87.5 |
4 | 1/16 | 6.25 | 93.75 |
5 | 1/32 | 3.125 | 96.875 |
6 | 1/64 | 1.156 | 98.844 |
n | 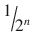 | 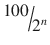 | 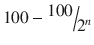 |
Loading Dose
If the time to reach a constant concentration by continuous or intermittent dosing is too long, a loading dose (LD) may be used to reach a greater constant concentration more quickly (Fig. 25.7). The calculation of the loading dose for a one-compartment model is:
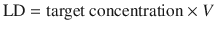 This is the principle underlying anaesthesia induction with propofol. Loading doses must be used cautiously, because they increase the likelihood of drug toxicity (e.g. hypotension with propofol).
This is the principle underlying anaesthesia induction with propofol. Loading doses must be used cautiously, because they increase the likelihood of drug toxicity (e.g. hypotension with propofol).
Dose calculations using a one-compartment model are not applicable to those drugs that are characterised using multi-compartment models. The use of V 1 results in a loading dose too high, while the use of V ss results in a loading dose too low. Too high a dose may cause transient toxicity, although slowing the rate of administration may prevent excessive concentrations during the distributive phase.
The time to peak effect (T peak) is dependent on clearance and effect site equilibration half-time (T 1/2 k eo). At a submaximal dose, T peak is independent of dose. At supramaximal doses, maximal effect will occur earlier than T peak and persist for longer duration because of the shape of the sigmoid E max response model. This is due to similar considerations described in time course of immediate effects. The T peak concept has been used to calculate optimal initial bolus doses [28], because V 1 and V ss poorly reflect the required scaling factor. A new parameter, the volume of distribution at the time of peak effect site concentration (V pe), is used and is calculated.
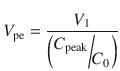
C 0 is the theoretical plasma concentration at t = 0 after the bolus dose, and C peak is the predicted effect site concentration at the time of peak effect site concentration. Loading dose can then be calculated as
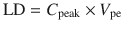
(25.10)
(25.11)
Population Modelling
The parameter estimates from the mathematical models used to analyse the data can be used to predict the time–concentration profiles of other doses. Attempts to predict what will happen in a further subject often become unstuck because a factor accounting for variability between subjects is missing. If the variability between patients is modelled, then it is possible to predict the magnitude of the difference between predictions and the observations in the next subject. There are three common approaches to modelling data collected from a group of subjects.
Naïve Pooled Data Approach
Time–concentration data are pooled together as if all d oses and all observations pertain to a single subject. Samples are taken at the same time in each individual. No information is available on individual subject profiles or parameters. This approach may be satisfactory if data are extensive for each subject and there is only minor between-subject variability, but may result in misrepresentation if data are few. Problems also arise interpreting results when data are missing from some subjects. No information can be gathered about the magnitude of between-subject variability and its causes.
Standard Two-Stage Approach
Individual profiles are analysed, and the individual structural parameters (e.g. V, CL) are then treated as variables and combined to achieve summary measures. Sampling times have greater flexibility but must be complete for each individual. If the estimates are not based on a similar number of measurements for each individual, or if the response in one individual is much more variable than another, some form of weighting is required. The between-subject variability can be estimated from the standard deviation of the individual estimates, but it is an overestimate of the true variability because each estimate also has variability due to imprecision of the estimate. It may be possible to identify covariates to explain some of the variability, but this does depend on having relatively good individual estimates of the parameters.
‘True’ Population Modelling
Population modelling using mixed-effects models [29, 30] has i mproved analysis and interpretation of PKPD data. Paediatric anaesthesiologists have embraced the population approach for investigating PK and PD. This approach provides a means to study variability in drug responses among individuals representative of those in whom the drug will be used clinically. Traditional approaches to interpretation of time–concentration profiles relied on ‘rich’ data from a small group of subjects. In contrast, mixed-effects models can be used to analyse ‘sparse’ (two to three samples) data from a large number of subjects. Sampling times are not crucial for population methods and can be fitted around clinical procedures or outpatient appointments. Sampling time bands rather than exact times are equally effective [31] and allow flexibility in children. Interpretation of truncated individual sets of data or missing data is also possible with this type of analysis, rendering it useful for paediatric studies. Population modelling also allows pooling of data across studies to provide a single robust PK analysis rather than comparing separate smaller studies that are complicated by different methods and analyses.
Mixed-effects models are ‘mixed’ because they describe the data using a mixture of fixed and random effects. Fixed effects predict the average influence of a covariate such as weight as an explanation of part of the between-subject variability in a parameter like clearance. Random effects describe the remaining variability between subjects that is not predictable from the fixed effect average. Explanatory covariates (e.g. age, size, renal function, sex, temperature) can be introduced that explain the predictable part of the between-individual variability. Nonlinear regression is performed by an iterative process to find the curve of best fit [32, 33].
Why Adult PK Parameters Do Not Work in Children
The use of adult parameter sets in TCI pumps for children results in concentrations lower than those observed in adults. A simple manual regimen for propofol infusion in adults [34] is of a bolus of 1 mg kg–1 followed by an infusion of 10 mg kg−1 h−1 (0–10 min), 8 mg kg−1 h−1 (10–20 min) and 6 mg kg−1 h−1 thereafter. Requirements for children, however, are greater. A loading dose of 3 mg kg−1 followed by an infusion rate of 15 mg kg−1 h−1 for the first 15 min, mg kg −1 h−1 from 15 to 30 min, 11 mg kg−1 h−1 from 30 to 60 min, 10 mg kg−1 h−1 from 1 to 2 h and 9 mg kg−1 h−1 from 2 to 4 h resulted in a steady-state target concentration of 3 mg L−1 in children 3–11 years [7]. Figure 25.3 shows that the adult ‘Marsh model’ predicts concentrations greater than all the paediatric model predictions, except that by Rigby-Jones et al. [15], unsurprising since the latter were derived from critically ill neonates! Increased requirements in children can be attributed to size factors. Decreased requirements in neonates are with consequent reduced clearance due to immature enzyme clearance systems. Organ dysfunction will also result in reduced requirements.
Major Paediatric PK Covariates
Growth and development are two major aspects of children not readily apparent in adults. How these factors interact is not necessarily easy to determine from observations because they are quite highly correlated. Drug elimination clearance, for example, may increase with weight, height, age, body surface area and creatinine clearance. One approach is to standardise for size before incorporating a factor for maturation [35].
Size
Clearance in children 1–2 years of age, expressed as L/h/kg, is commonly greater than that observed in older children and adolescents. This is a size effect and is not due to bigger livers or increased hepatic blood flow in that subpopulation. This ‘artefact of size’ disappears when allometric scaling is used to replot the same data. Allometry is a term used to describe the nonlinear relationship between size and function. This nonlinear relationship is expressed as
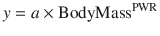
where y is the variable of interest (e.g. basal metabolic rate), a is the allometric coefficient and PWR is the allometric exponent. The value of PWR has been the subject of much debate. Basal metabolic rate (BMR) is the commonest variable investigated, and camps advocating for a PWR value of 2/3 (i.e. body surface area) are at odds with those advocating a value of 3/4.
(25.12)
Support for a value of 3/4 comes from investigations that show the log of basal metabolic rate (BMR) plotted against the log of body weight produces a straight line with a slope of 3/4 in all species studied, including humans. Fractal geometry is used to mathematically explain this phenomenon. The 3/4 power law for metabolic rates was derived from a general model that describes how essential materials are transported through space-filled fractal networks of branching tubes [36]. A great many physiological, structural, and time-related variables scale predictably within and between species with weight (W) exponents (PWR) of 3/4, 1 and 1/4, respectively [37].
These exponents have applicability to pharmacokinetic parameters such as clearance (CL exponent of 3/4), volume (V exponent of 1), and half-time (T 1/2 exponent of 1/4) [37]. The factor for size (F size) for total drug clearance may be expressed as
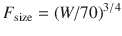
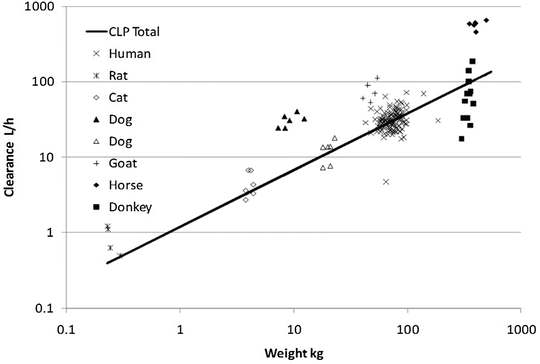
Clearance is a metabolic function, and while the process for drug clearance may differ between animals, the clearance of tramadol plotted against the log of body weight produces a straight line with a slope of 3/4 in all species studied (Fig. 25.8) [38].
(25.13)
Fig. 25.8
Weight-predicted tramadol total clearance (CLP total) compared to human allometric prediction using a 3/4 power exponent (solid line) (Reproduced From Holford S, Allgaert K, Anderson BJ, Kukanich B, Sousa AB, Steinman A, Pypendop BH, Mehvar M, Giorgi M, Holford NH. Parent-metabolite pharmacokinetic models for tramadol—tests of assumptions and predictions. J Pharmacol Clin Toxicol 2014;2(1):1023 [38], under a Creative Commons Attribution Licence. https://www.jscimedcentral.com/Pharmacology/pharmacology-spidpharmacokinetics-1023.pdf)
Maintenance dose is determined by clearance. The difference in drug clearance between an adult and a child is predictable from weight using theory-based allometry:
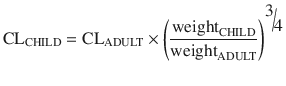
Allometric theory predicts maintenance dose per kg is higher in children. For example, remifentanil clearance is increased in neonates, infants and children when expressed as per kilogram [22]. However, remifentanil clearance in children 1 month–9 years is similar to adult rates when scaled using an allometric exponent of 3/4 [23]. Nonspecific blood esterases that metabolise remifentanil are mature at birth [39], and that is when clearance (L/h/kg) is highest.
(25.14)
Maturation
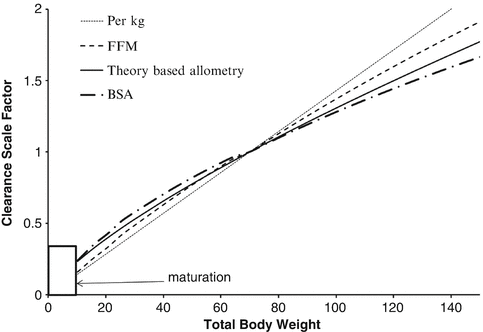
Unlike remifentanil clearance , allometry alone is insufficient to predict clearance in neonates and infants from adult estimates for most drugs (Fig. 25.9) [40, 41]. The addition of a model describing maturation is required. The sigmoid hyperbolic or Hill model [42] has been found useful for describing this maturation factor (MF).
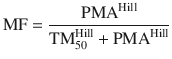
The TM50 describes the maturation half-time, while the Hill coefficient relates to the slope of this maturation profile. Maturation of clearance begins before birth, suggesting that postmenstrual age (PMA) would be a better predictor of drug elimination than postnatal age (PNA) [37]. Figure 25.10 shows the maturation profile for dexmedetomidine expressed as both the standard per kilogram model and using allometry. Clearance is immature in infancy. Clearance is greatest at 2 years of age, decreasing subsequently with age. This ‘artefact of size’ disappears with use of the allometric model.
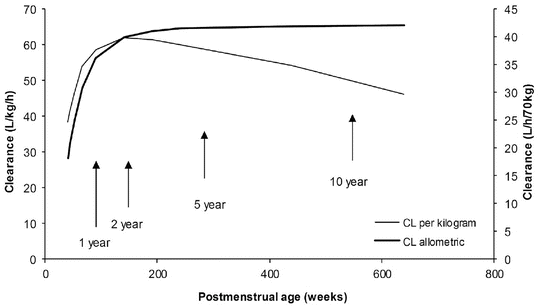
Fig. 25.9
Body size metrics used to describe clearance changes with weight for individuals of average height for weight. The clearance scale factor shows how clearance would differ with weight. A nonlinear relationship exists between weight and clearance using theory-based allometry. The per kg method increasingly overestimates clearance in adults and underestimates clearance in children. The BSA method overestimates clearance in children compared to theory-based allometry. Scaling with fat-free mass (FFM) lies between the per kg method and theory-based allometry. An additional function is required to describe maturation (Reproduced from Anderson BJ, Holford NH. Understanding dosing: children are small adults, neonates are immature children. Arch Dis Child 2013; 98: 737–44 [2] with kind permission of the BMJ Publishing Group)
(25.15)
Fig. 25.10
The clearance maturation profile of dexmedetomidine expressed using the per kilogram model and the allometric 3/4 power model. This maturation pattern is typical of many drugs cleared by the liver or kidneys (Data adapted from Potts AL, Anderson BJ, Warman GR, Lerman J, Diaz SM, Vilo S. Dexmedetomidine pharmacokinetics in pediatric intensive care—a pooled analysis. Pediatr Anesth 2009;19:1119–29 [624], with kind permission from John Wiley and Sons)
Organ Function
Changes associated with normal growth and development can be distinguished from pathological changes describing organ function (OF) [35]. Morphine clearance is reduced in neonates because of immature glucuronide conjugation, but clearance was lower in critically ill neonates than healthier cohorts [43–45], possibly attributable to reduced hepatic function. The impact of organ function alteration may be concealed by another covariate. For example, positive pressure ventilation may be associated with reduced clearance rather than intensive care admission. This effect may be attributable to a consequent reduced hepatic blood flow with a drug that has perfusion-limited clearance (e.g. propofol, morphine).
Pharmacokinetic parameters (P) can be described in an individual as the product of size (F size), maturation (MF) and organ function (OF) influences, where P std is the value in a standard size adult without pathological changes in organ function [35]:
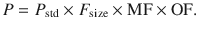
This methodology is increasingly used to describe clearance changes with age [46]. An understanding of these principles can be used to predict dose in children using target concentration methodology [2].
(25.16)
When maturation changes have not been described using real data, then an alternative method known as physiological-based pharmacokinetic (PBPK) models can be used to predict changes with age. Organ maturation, body composition and ontogeny of drug elimination pathways have marked effects on pharmacokinetic parameters in the first few years of life. PBPK models require detailed physiological data. Data on ontogeny of individual clearance pathways, derived from measurements of enzyme expression and activity in postmortem livers and from in vivo data from drugs that are cleared by similar pathways, are useful. Continued input of information concerning genetic, physiological, organ and tissue size and composition, protein binding, demographic and clinical data into the library and algorithms for PBPK modelling programmes has progressively improved their prediction ability. These models have been used to assist with first-time dosing in children [40, 41, 47]. The introduction of population variability in enzyme abundance and activity contributes to between-individual variability estimates [48]. This approach has been recently used to investigate fentanyl maturation changes with age in neonates [49] .
Dosing in Obese Children
Volume (determining loading dose) and clearance (determining maintenance dose) of some drugs are known to be changed in obesity [50]. Although body fat has minimal metabolic activity, fat mass contributes to overall body size and may have an indirect influence on both metabolic and renal clearance. On the other hand, the volume of distribution of a drug depends on its physicochemical properties [51]. There are drugs whose apparent volume of distribution may be independent of fat mass (e.g. digoxin) or be extensively determined by it (e.g. diazepam). A number of size descriptors (Fig. 25.9) have been put forward for use in the obese patient, e.g. total body weight (TBW), lean body weight (LBW), ideal body weight (IBW), body mass index (BMI), fat-free mass (FFM) and normal fat mass (NFM).
These size descriptors invariably demonstrate nonlinear relationships between weight and clearance. The best size descriptor accounting for obesity remains unknown [52]. LBW is often advocated for use in obese, but that descriptor may not apply for all drugs. An infusion of propofol is commonly used for paediatric anaesthesia. Infusion rate is dependent on clearance, and an incorrect estimate of clearance may lead to inadequate anaesthesia and awareness. Propofol clearance in obese children [53] and adults [54] and nonobese adults and children [55, 56] is best predicted using TBW as the size descriptor with theory-based allometry.
However, TBW may be inappropriate for remifentanil where lean body weight appears to be a better size descriptor [57]. The use of normal fat mass (NFM) [58] with allometric scaling as a size descriptor may prove versatile [59–62]. That size descriptor uses the idea of fat-free mass (similar to LBW but excludes lipids in cell membranes, CNS and bone marrow) plus a ‘bit more’. The ‘bit more’ will differ for each drug, and the maximum ‘bit more’ added to fat-free mass would equal TBW.
![$$ \mathrm{F}\mathrm{F}\mathrm{M}={\mathrm{WHS}}_{\max}\times {\mathrm{HT}}^2\times \left[\raisebox{1ex}{$\mathrm{T}\mathrm{B}\mathrm{W}$}\!\left/ \!\raisebox{-1ex}{$\left({\mathrm{WHS}}_{50}\times {\mathrm{HT}}^2+\mathrm{T}\mathrm{B}\mathrm{W}\right)$}\right.\right] $$](/wp-content/uploads/2017/07/A339434_1_En_25_Chapter_Equ17.gif)
where WHSmax is the maximum FFM for any given height (HT, m) and WHS50 is the TBW value when FFM is half of WHSmax. For men, WHSmax is 42.92 kg m−2 and WHS50 is 30.93 kg m−2, and for women WHSmax is 37.99 kg m−2 and WHS50 is 35.98 kg m−2 [63].
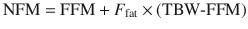
The parameter F fat is estimated and accounts for different contributions of fat mass. If F fat is estimated to be zero, then FFM alone predicts size, while if F fat is 1, then size is predicted by TBW. This parameter is drug specific and also specific to the PK parameter such as clearance or volume of distribution. It has a value of 0.211 for GFR which implies that 21 % of fat mass is a size driver for kidney function in addition to FFM [59]. Size based on NFM assumes that FFM is the primary determinant of size with an extra F fat factor (which may be positive or negative) that determines how fat mass contributes to size. A negative value for F fat might suggest organ dysfunction, not an uncommon scenario in the morbidly obese.
(25.17)
(25.18)
Pharmacodynamic Models
Pharmacokinetics is what the body does to the drug, while pharmacodynamics is what the drug does to the body. The precise boundary between these two processes is ill defined and often requires a link describing movement of drug from the plasma to the effect site and its target. Drugs may exert effect at nonspecific membrane sites, by interference with transport mechanisms, by enzyme inhibition or induction or by activation or inhibition of receptors.
Minimal Effective Concentration
The minimal effective analgesic concentration can sometimes be determined by titration of an analgesic to achieve satisfactory pain at rest or with stimulus. Blood assay for analgesic drug concentration at these times can be used to determine an effective concentration. Reassessment when pain recurs or after further analgesic administration improves accuracy of assessment. This technique has been used with to determine the minimal effective analgesic concentration for oxycodone [64].
Sigmoid E max Model
A better understanding of drug effect is achieved if response over a broad concentration range is explored. The relation between drug concentration and effect may be described by the Hill equation (for sigmoid E max model, see maturation model above) [42].
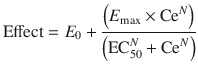
where E 0 is the baseline response, E max is the maximum effect change, Ce is the concentration in the effect compartment, EC50 is the concentration producing 50 % E max and N is the Hill coefficient defining the steepness of the concentration–response curve (Fig. 25.11). Efficacy is the maximum response on a dose or concentration–response curve. EC50 can be considered a measure of potency relative to another drug provided N and E max for the two drugs are the same. This model has been used to describe propofol [53, 65] and remifentanil [66] effect in children.
(25.19)
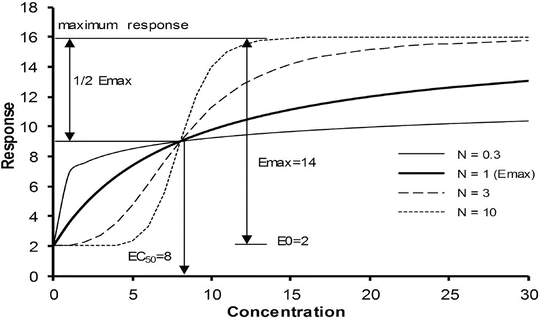
Fig. 25.11
The sigmoid E max model is commonly used to describe the relationship between drug response and concentration. Changing the Hill coefficient dramatically alters the shape of the curve
Quantal Effect Model
The potency of anaesthetic vapours may be expressed by minimal alveolar concentration (MAC) , and this is the concentration at which 50 % of subjects move in response to a standard surgical stimulus. MAC appears at first sight to be similar to EC50 but is an expression of quantal response rather than magnitude of effect. There are two methods of estimating MAC. Responses can be recorded over the clinical dose range in a large number of subjects and logistic regression applied to estimate the relationship between dose and quantal effect; the MAC can then be interpolated. Large numbers of subjects may not be available, and so an alternative is often used. The ‘up and down’ method described by Dixon [67, 68] estimates only the MAC rather than the entire sigmoid curve. It involves a study of only one concentration in each subject, and, in a sequence of subjects, each receives a concentration depending upon the response of the previous subject; the concentration is either increased if the previous subject did not respond or decreased if they did (Fig. 25.12). The MAC is usually calculated either as the mean concentration of equal numbers of responses and no responses or is the mean concentration of pairs of ‘response–no response’.
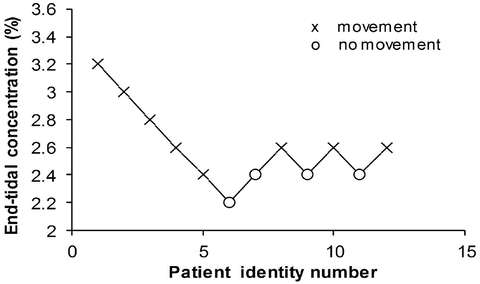
Fig. 25.12
Schematic of the ‘up and down’ method to estimate MAC. It involves a study of only one concentration in each subject, and, in a sequence of subjects, each receives a concentration depending upon the response of the previous subject
This method has also been used for drugs other than inhalation vapours. In order to determine the dexmedetomidine dose that could be given as a rapid 5 s bolus to healthy children during TIVA without causing significant haemodynamic effects, children were given dexmedetomidine, starting at 0.3 mcg/kg with 0.1 mcg/kg intervals. The dose that had no haemodynamic response in half the subjects was 0.5 mcg/kg [69].
Logistic Regression Model
When the pharmacological effect is difficult t o grade, then it may be useful to estimate the probability of achieving the effect as a function of plasma concentration. Effect measures such as movement/no movement or rousable/non-rousable are dichotomous. Logistic regression is commonly used to analyse such data, and the interpolated EC50 value refers to the probability of response. For example, an EC50 of 0.52 mg/L for arousal after ketamine sedation in children has been estimated using this technique [70].
Linking PK with PD
A simple situation in which drug effect is directly related to concentration does not mean that drug effects parallel the time course of concentration. This occurs only when the concentration is low in relation to EC50. In this situation the half-life of the drug may correlate closely with the half life of drug effect. Observed effects may not be directly related to serum concentration. Many drugs have a short half-life but a long duration of effect. This may be attributable to induced physiological changes (e.g. aspirin and platelet function) or may be due to the shape of the E max model. If the initial concentration is very high in relation to the EC50, then drug concentrations 5 half-lives later, when we might expect minimal concentration, may still exert considerable effect (Fig. 25.5). There may be a delay due to the transfer of the drug to effect site (NMBD), a lag time (diuretics), a physiological response (antipyresis), an active metabolite (propacetamol) or a synthesis of physiological substances (warfarin).
A plasma concentration–effect plot can form a hysteresis loop because of this delay in effect (Fig. 25.13). Hull et al. [71] and Sheiner et al. [72] introduced the effect compartment concept for muscle relaxants. A single first-order parameter (T 1/2 k eo) describes the equilibration half-time (Fig. 25.2).
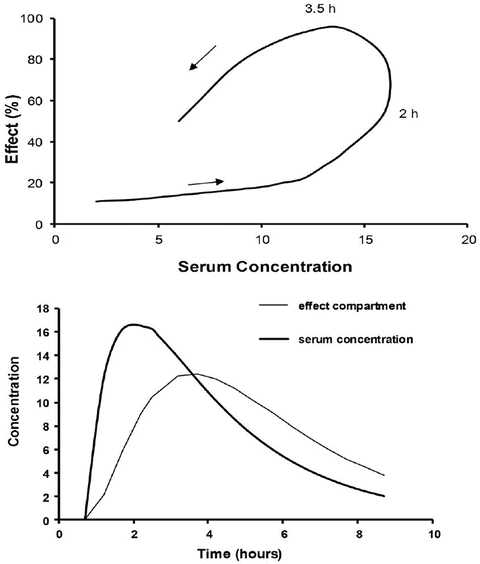
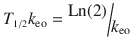
This mathematical trick assumes concentration in the central compartment is the same as that in the effect compartment at equilibration but that a time delay exists before the drug reaches the effect compartment. The concentration in the effect compartment is used to describe the concentration–effect relationship [73].
Fig. 25.13
The counterclockwise hysteresis loop observed after an orally administered drug is shown in the upper panel. Effect increases between 2 and 3.5 h even though serum concentration is decreasing. The lower panel shows that the time–concentration profile for an effect compartment is delayed compared to that in the serum
(25.20)
Adult T 1/2 k eo values are well described, e.g. morphine 16 min, fentanyl 5 min, alfentanil 1 min and propofol 3 min. This T 1/2 k eo parameter is commonly incorporated into target-controlled infusion (TCI) pumps in order to achieve a rapid effect site concentration.
A T 1/2 k eo for propofol in children determined by simultaneous PK–PD modelling is uncommonly described. An estimate of 1.2 min (95 %CI 0.85–2.1) has been reported in obese children [53]. We might expect a shorter T 1/2 k eo with decreasing age based on size models [74] Faster half-times in children can be accounted for by considering physiologic time that scales to a power of 1/4 [75].
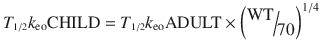
A decreasing T 1/2 k eo with age (linked to weight) has been described for propofol in children [65]. Similar results have been demonstrated for sevoflurane and BIS [76]. If unrecognised, this will result in excessive dose in a young child if the effect site is targeted and peak effect is anticipated to be later than it actually is because it was determined in a teenager or adult.
(25.21)
What Is a PK–PD Model?
When both PK and PD data are collected simultaneously and parameters for both models are estimated together, then the model is described as ‘integrated’. PK estimates should not be used in conjunction with PD estimates taken from a different data set without a few ‘fudge factors’. T peak methodology (Eqs. (25.10) and (25.11)) is commonly used to estimate T 1/2 k eo that then links separate PK and PD data sets. Model dependence of the T 1/2 k eo was demonstrated by an estimate of 1.7 min with the Kataria et al. [8] parameter set and 0.8 min with the Paedfusor® (Graseby Medical Ltd., Hertfordshire, United Kingdom) parameter set [14]. These estimates are similar to that estimated in obese children using PK–PD modelling [53].
Paediatric Pharmacokinetic Considerations
Distribution
At its simplest, the volume of distribution determines the initial dose of a drug. It is a scaling factor. Distribution is influenced by body composition, protein binding, haemodynamics (e.g. regional blood flow) and membrane permeability.
Body Composition
Total body water and extracellular fluid (ECF) [77] are increased in neonates, and reduction tends to follow postnatal age (PNA). Polar drugs such as the non-depolarising neuromuscular blocking drugs (NMBDs) and aminoglycosides distribute rapidly into the ECF, but enter cells more slowly. The initial dose of such drugs is consequently higher in the neonate compared to the infant, older child or adult.
The percentage of body weight contributed by fat is 3 % in a 1.5 kg premature neonate and 12 % in a term neonate; this proportion doubles by 4–5 months of age. ‘Baby fat’ is lost when infants start walking and protein mass increases (20 % in a term neonate, 50 % in an adult). These body component changes affect volumes of distribution of drugs. Volume of distribution influences initial dose estimates. Fentanyl has an increased volume of distribution in neonates. The volume of distribution at steady state is 5.9 (SD 1.5) L/kg in a neonate under 1 month of age compared to 1.6 (SD 0.3) L/kg in an adult [78]. This may contribute to the reduced degree of respiratory depression seen after single doses as high as 10 mcg/kg in older term neonates. The dramatic increase in muscle bulk in children from 3 years until adolescence influences drug dose required for neuromuscular blockade. The ED95 of vecuronium, for example, is 47 SD 11 mcg/kg in neonates and infants, 81 SD 12 mcg/kg in children between 3 and 10 years of age and 55 SD 12 mcg/kg in patients aged 13 years or older [79]. Dose is greater than anticipated in neonates who have immaturity of the neuromuscular junction because the ECV is increased, but the duration of neuromuscular blockade is greater in neonates because of immature clearance pathways. The plasma concentration required in neonates to achieve the same level of neuromuscular block as in children or adults is 20–50 % less [80].
Reduction of propofol concentrations after induction is attributable to redistribution rather than rapid clearance because its pharmacokinetics is described using more than one compartment. Neonates have low body fat and muscle content, and so less propofol is apportioned to these tissues. Delayed awakening occurs because CNS concentration remains higher than that observed in older children as a consequence of reduced redistribution.
Plasma Proteins
Albumen and alpha-1 acid glycoprotein (AAG) concentrations (Fig. 25.14) are reduced in neonates but are similar to those in adults by 6 months, although between-patient variability is high (e.g. AAG 0.32–0.92 g/L) [81, 82]. Bupivacaine is bound to AAG. The recommended bolus epidural dose of bupivacaine in neonates is lower than in children (1.5–2 mg/kg vs. 2.5 mg/kg) because a greater proportion will be unbound drug and it is unbound drug that exerts effect. AAG is an acute-phase reactant that increases after surgical stress. This causes an increase in total plasma concentrations for low to intermediate extraction drugs such as bupivacaine [83]. The unbound concentration, however, will not change because clearance of the unbound drug is affected only by the intrinsic metabolising capacity of the liver. Any increase in unbound concentrations observed during long-term epidural is attributable to reduced clearance rather than AAG concentration [84, 85].
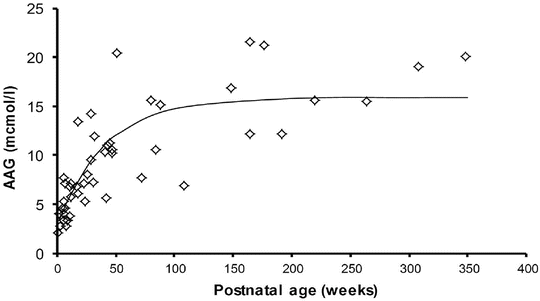
Fig. 25.14
Alpha-1 acid glycoprotein changes with age. Adapted from Booker P. Br J Anaesth 1996;76:365–8
Plasma albumin concentrations are lowest in premature infants, and other foetal proteins such as alpha-fetoprotein (synthesised by the embryonic yolk sac, foetal gastrointestinal tract and liver that has 40 % homology with albumin) have reduced affinity for drugs. In addition, increased concentrations of free fatty acids and unconjugated bilirubin compete with acidic drugs for albumin binding sites. Neonates also have a tendency to manifest a metabolic acidosis that alters ionisation and binding properties of plasma proteins. Serum albumin concentrations approximate adult values by 5 months of age, and binding capacity approaches adult values by 1 year of age. The induction dose of thiopentone is lower in neonates than older children. It is possible that this is related to decreased binding of thiopentone to plasma albumin; 13 % of the drug is unbound in newborns compared to 7 % in adults [86].
Regional Blood Flows
The initial phase of distribution after intravenous administration reflects regional blood flow. Consequently, the brain, heart and liver are the tissues first exposed to the drug. Drug is then redistributed to other relatively well-perfused tissues, such as the skeletal muscle. There is a much slower tertiary distribution to relatively underperfused tissues of the body that is noted with long-term drug infusions. These changes contribute to a shorter context-sensitive half-time in infants with quicker ‘awakening’ after sedative drugs; these infants have less fat and muscle bulk that the drug can redistribute to and later leach out from. Clearance, however, is typically reduced in neonates and contributes to the longer observed context-sensitive half-time.
Apart from the neonatal circulatory changes that occur at birth (e.g. secondary to functional closure of the ductus venosus and ductus arteriosus), there are differences in relative organ mass and regional blood flow change with growth and development during the first few months of life. Blood flow, relative to cardiac output, to the kidney and brain increases, while that to the liver decreases through the neonatal period [87]. Cerebral and hepatic mass as a proportion of body weight are much higher in the infant than in the adult [88].
Mean cerebral blood flow is highest in early childhood (70 mL/min/100 g) at about 3–8 years of age [89]. It is reduced before this age in neonates and later in adults, where flows are similar (50 mL/min/100 g) [90]. The highly lipophilic induction agents diffuse rapidly across the blood–brain barrier to achieve concentration equilibrium with brain tissue. Reduced cardiac output in neonates and reduced cerebral perfusion mean that the onset time after intravenous induction is slower in neonates that in early childhood. Offset time is also delayed because redistribution to the well-perfused and deep, underperfused tissues is less.
Blood–Brain Barrier (BBB)
The BBB is an elaborate network of complex tight junctions between specialised endothelial cells that restricts the paracellular diffusion of hydrophilic molecules from the blood to the brain substance. Confusion over the importance of this barrier in the neonate exists, partly because of early studies comparing respiratory depression caused by the opioids, morphine and pethidine. Greater respiratory depression was evident in neonates after morphine given as an adult equipotent dose of pethidine [91]. This finding is consistent with pethidine, unlike morphine, being lipid soluble and therefore crossing the immature or mature BBB equally [91]. However, plasma opioid concentrations were not measured in that study, and the increased neonatal respiratory depression observed after morphine when given the same dose (mg/kg) as adults could be due to reduced volume of distribution of morphine in term neonates 1–4 days (1.3 L/kg) compared to those at 8–60 days (1.8 L/kg), 61–180 days (2.4 L/kg) and adults (2.8 L/kg) [43]. Consequently we might expect initial concentrations of morphine to be higher in neonates than in adults and consequent respiratory depression greater. Respiratory depression, as measured by carbon dioxide response curves or by arterial oxygen tension, is similar in children from 2 to 570 days of age at the same morphine concentration [92].
The BBB may have impact in other ways. There are specific transport systems selectively expressed in the barrier endothelial cell membranes that mediate the transport of nutrients into the CNS and of toxic metabolites out of the CNS. Small molecules access foetal and neonatal brains more readily than they do adult brains [93]. BBB function improves gradually throughout foetal brain development, possibly reaching maturity at term [93]. Kernicterus , for example, is more common in the premature neonate than the term neonate. Pathological conditions within the CNS can cause BBB breakdown, or alterations in transport systems play an important role in the pathogenesis of many CNS diseases. Proinflammatory substances and specific disease-associated proteins often mediate BBB dysfunction [94].
Fentanyl is actively transported across the BBB by a saturable ATP-dependent process, while ATP-binding cassette proteins such as P-glycoprotein actively pump out opioids such as fentanyl and morphine [95]. P-glycoprotein modulation significantly influences opioid brain distribution and onset time, magnitude and duration of analgesic response [96]. Modulation may occur during disease processes, increased temperature or other substances (e.g. verapamil, magnesium) [95]. Genetic polymorphisms affecting P-glycoprotein-related genes may explain some individual differences in CNS-active drug sensitivity [97].
Absorption
Absorption characteristics will impact on amount of drug available, maximum concentration, speed of onset of effect, duration of effect and time to offset of effect.
Enteral
The rate at which most drugs are absorbed when given by the oral route is slower in neonates than in older children because gastric emptying is delayed and normal adult rates may not be reached until 6–8 months [98–101]. Slow gastric emptying and reduced clearance may dictate both reduced doses and reduced frequency of administration (Fig. 25.15). This has been demonstrated for both cisapride [102] and acetaminophen [103].
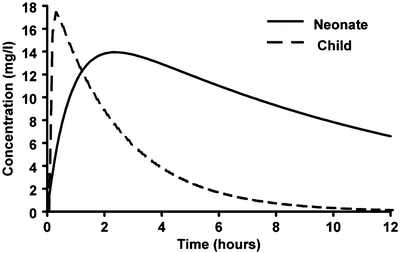
Fig. 25.15
Simulated mean predicted time–concentration profiles for a term neonate, a 1-year-old infant and a 5-year-old child given paracetamol elixir. The time to peak concentration is delayed in neonates due to slow gastric emptying and reduced clearance (Reproduced from Anderson BJ, van Lingen RA, Hansen TG, Lin YC, Holford NH. Acetaminophen developmental pharmacokinetics in premature neonates and infants: a pooled population analysis. Anesthesiology 2002; 96: 1336–45 [103], with kind permission of Wolters Kluwer Health)
Delayed gastric emptying and reduced clearance may dictate reduced doses and frequency of repeated drug administration. For example, a mean steady-state target paracetamol concentration greater than 10 mg/L at trough can be achieved by an oral dose of 25 mg/kg/day in preterm neonates at 30 weeks, 45 mg/kg/day at 34 weeks and 60 mg/kg/day at 40 weeks PMA [103]. Because gastric emptying is slow in preterm neonates, dosing may only be required twice a day [103].
Intramuscular
The intramuscular route is commonly frowned upon in children. It retains high bioavailability, but absorption is delayed compared to the intravenous route. Ketamine however remains popular, and peak concentrations are reached within 10 min after 4 mg/kg [106]. Dexmedetomidine has a similar absorption profile [107, 108].
Nasal
Exploration of alternative delivery routes in young children has centred on the nasal passages [109]. Nasal diamorphine 0.1 mg/kg is used in the UK for forearm fracture pain in the emergency room [110–113]. It is rapidly absorbed as a nasal spray (0.1 mg/kg) in 0.2 mL sterile water, with peak morphine plasma concentrations (T peak) occurring at 10 min [113]. Nasal S-ketamine 2 mg/kg results in peak plasma concentrations of 355 ng/mL within 18 min. Nasal fentanyl (150 mcg/mL) 1.5 mcg/kg given to children (3–17 years) for fracture pain resulted in good analgesia. Peak concentrations were at 13 min [114, 115]. Similar results are reported for nebulised fentanyl (4 mcg/kg) given through a standard nebuliser [116]. Flumazenil concentrations peak within a few minutes after nasal administration [117], while midazolam takes approximately 12 min [118]. Dexmedetomidine is somewhat slower and peak concentrations were not reached until 38 min [119]. Clonidine administered as nasal aerosol (3–8 mcg/kg) was not found to achieve adequate preoperative sedation within 30 min of administration [120], attributable to slow absorption (T peak 1.5–3 h) [121]. There remain concerns that intranasal drugs may pass through the posterior nasopharynx or irritate the vocal cords [122].
Advances in aerosol delivery devices have improved dosing accuracy. Administration of ketorolac 15 mg (weight <50 kg) or 30 mg (weight >50 kg) by the intranasal route resulted in a rapid increase in plasma concentration (time to peak concentration was 52 SD 6 min) and may be a useful therapeutic alternative to IV injection. A target concentration of 0.37 mg/L in the effect compartment was achieved within 30 min and remained above that target for 10 h [123]. The nasal passages change with age, and so it would be unsurprising if absorption by that route did not also change with age.
Cutaneous
The larger relative skin surface area, increased cutaneous perfusion and thinner stratum corneum in neonates [124] increase absorption and exposure of topically applied drugs (corticosteroids, local anaesthetic creams, antiseptics). Neonates have a tendency to form methaemoglobin because they have reduced levels of methaemoglobin reductase and foetal haemoglobin is more readily oxidised compared to adult haemoglobin. This, combined with increased absorption through the neonatal epidermis, resulted in reluctance to use lidocaine–prilocaine cream for repeat use in this age group [125, 126].
Alveolar
Anaesthetic delivery to the alveoli is determined largely by alveolar ventilation and functional residual capacity (FRC) . Neonates have increased alveolar ventilation. They also have a smaller FRC compared to adults because of increased chest wall compliance; this causes an increase in the speed of delivery. Pulmonary absorption is generally more rapid in infants and children than in adults [127]. The greater cardiac output and greater fraction of the cardiac output distributed to the vessel-rich tissue group (i.e. a clearance factor) and the lower tissue/blood solubility (i.e. a volume factor) also affect the more rapid wash-in of inhalational anaesthetics in the younger age group [128]. Solubility determines volume of distribution. An inhalational agent with a greater volume of distribution will take longer to reach a steady-state concentration when delivered at a constant rate. The solubility in blood of halothane, isoflurane, enflurane and methoxyflurane is 18 % less in neonates than in adults [129], attributable to altered serum albumin, globulin, cholesterol and triglyceride concentrations. The solubility of these same agents in the vessel-rich tissue group in neonates is approximately one half of those in adults [129]. The latter may be due to the greater water content and decreased protein and lipid concentration in neonatal tissues. Infants, with their decreased solubility would be expected to have a shorter time to reach a predetermined F E /F I ratio because of a smaller volume of distribution. Age has little effect on the solubility of the less soluble agents, nitrous oxide and sevoflurane [130].
Induction of anaesthesia may be slowed by right-to-left shunting of blood in neonates suffering cyanotic congenital cardiac disease or intrapulmonary conditions. This slowing is greatest with the least soluble anaesthetics. Left-to-right shunts usually have minimal impact on uptake because cardiac output is increased so that systemic tissue perfusion is maintained at normal levels. The flow of mixed venous blood returning to the right heart ready for anaesthetic uptake is normal. If cardiac output is not increased, and peripheral perfusion is reduced, then there will be less anaesthetic uptake in the lung. Although alveolar anaesthetic partial pressure may be observed to rise rapidly, there is a slower rise in tissue partial pressure and anaesthetic effect is delayed.
Bioavailability
The oral bioavailability may be affected by interactions with food when feeding is frequent in the neonate (e.g. phenytoin [131]), by the use of adult formulations that are divided or altered for paediatric use (nizatidine [132]) and by lower cytochrome P450 enzyme activity in the intestine. The latter may cause an increased bioavailability of midazolam because CYP3A activity is reduced [133]. The use of adult vials for paediatric use may result in dose inaccuracy, causing a relative increase or decrease in assumed bioavailability [134].
Analgesic medications and delivery systems commonly used in adults may not be possible or practicable in children because they do not have behavioural maturity. Infants are unable to use patient-controlled analgesia devices. Dose accuracy is lost when buccal and sublingual administration is attempted because those routes require prolonged exposure to the mucosal surface. Infants find it difficult to hold drug in their mouth for the requisite retention time (particularly if taste is unfavourable), and this results in more swallowed drug or drug spat out than in adults [135]. If the drug has a high first-pass effect, then the lower relative bioavailability results in lower plasma concentrations. Although many analgesics are available in an oral liquid formulation, taste is a strong determinant of compliance and unpalatable preparations may be refused [136]. Taste preferences change with age.
First-pass effect impacts on bioavailability and contribution of active metabolites to effect. The oral bioavailability of clonidine is low (F = 0.55) in children 3–10 years. Consequently, higher oral doses of clonidine (per kg) are required when this formulation is used to achieve concentrations similar to those reported in adults [137]. Oral absorption is slow (absorption half-time 0.45 h) and peak concentrations not reached until 1 h. Similarly, oral ketamine needs to be given in doses of up to 10 mg/kg to achieve therapeutic effect in children 1–8 years suffering burns [138]. Not only was bioavailability reduced (F = 0.45) but absorption was also slow; absorption half-time was 59 min and had high between-subject variability in this cohort [138]. Analgesic effect , however, may be contributed by the increased concentration of the active metabolite norketamine.
The frequent passage of stools in the neonate may render suppository use ineffective. Variable absorption and bioavailability have resulted in respiratory arrest when repeat opioids are administered by the rectal route to children [139].
Metabolism and Excretion
Hepatic Metabolism
Hepatic metabolic reactions are categorised as phase I (non-synthetic) or phase II (synthetic). Phase I reactions include oxidation, reduction and hydrolysis, while phase II processes involve conjugation with other molecules, notably glucuronide, glycine and sulphate. These water-soluble metabolites are excreted by the kidneys.
Hepatic drug metabolism activity appears as early as 9–22 weeks’ gestation when foetal liver enzyme activity may vary from 2 to 36 % of adult activity [140–142]. These pathways then develop at different rates. Microsomal enzyme activity can be classified into three groups: (1) mature at birth but decreasing with age (e.g. CYP3A7 responsible for methadone clearance in neonates [143]), (2) mature at birth and sustained through to adulthood (e.g. plasma esterases that clear remifentanil [39]) and (3) immature at birth [144].
Medications that are extensively metabolised by the liver or other organs (e.g. the intestines or lungs) are referred to as having high extraction ratios (perfusion-limited clearance, e.g. propranolol, morphine and midazolam). This extensive metabolism produces a first-pass effect in which a large proportion of an enteral dose is inactivated as it passes through the organ before reaching the systemic circulation. Other drugs with low intrinsic clearance (e.g. aspirin, diazepam) are known as having capacity-limited clearance.
Cytochromes P450: Phase I Reactions
Cytochromes P450 are heme-containing proteins that provide most of the phase I drug metabolism for lipophilic compounds in the body. The generally accepted nomenclature of the cytochrome P450 isozymes begins with CYP and groups enzymes with more than 36 % DNA homology into families designated with an Arabic number followed by alphanumeric letters for the subfamily of closely related proteins (>77 % homology) followed by a number for the specific enzyme gene, such as CYP3A4 [145–147]. Isozymes that are important in human drug metabolism are found in the CYP1, CYP2 and CYP3 gene families. Table 25.3 outlines the P450 isozymes and their common substrates. This table also outlines enzyme activity or enzyme concentration in the liver and changes with age; these are not the same as total clearance, which is determined by enzyme activity, hepatic size and blood flow. Clearance changes with age for many P450 enzyme processes remain poorly described. There are also both genetic and ethnic polymorphisms leading to clinically important differences in the capacity to metabolise drugs, and these differences can make individual drug responses in some cases unpredictable.
Table 25.3
Developmental patterns and activities for P450 enzymes in the neonate
Enzyme | Substrates | Inducers | Inhibitors | Developmental changes |
|---|---|---|---|---|
CYP1A2 | Acetaminophen, caffeine, theophylline, warfarin | Cigarette smoke, charcoal-broiled meat, omeprazole, cruciferous vegetables | α-Naphthoflavone | Not active to an appreciable extent in human foetal liver, but adult concentrations reached by 4 months of age |
CYP2A6 | Coumarin, nicotine | Barbiturates | Tranylcypromine | |
CYP2C9 | Diclofenac, phenytoin, torsemide, S-warfarin tolbutamide, ibuprofen | Rifampin | Sulfaphenazole, sulfinpyrazone | Low activity in neonate |
CYP2C19 | Phenytoin, diazepam, omeprazole, propranolol | Rifampin, phenobarbital | Tranylcypromine, cimetidine | Rapid maturation in the first week of life, with adult activity reached by 6 months of age |
CYP2D6 | Amitriptyline, captopril, codeine, dextromethorphan, fluoxetine, hydrocodone, ondansetron, propafenone, propranolol, timolol | None known | Fluoxetine, quinidine, cimetidine | Low to absent in foetal liver but uniformly present at 1 week of postnatal age. Poor activity (approximately 20 % of adult values) at 1 month of postnatal age |
CYP3A4 | Acetaminophen, alfentanil, amiodarone, budesonide, carbamazepine, diazepam, erythromycin, lidocaine, midazolam, nifedipine, omeprazole, cisapride, theophylline, verapamil, R-warfarin, oxycodone | Carbamazepine, dexamethasone, phenobarbital, phenytoin, rifampin | Azole antifungals, ethinyl estradiol, naringenin, troleandomycin, erythromycin | CYP3A4 has low activity in the first month of life, with approach towards adult levels by 6–12 months postnatally |
CYP3A7 | Dehydroepiandrosterone, ethinyl estradiol, various dihydropyrimidines | Carbamazepine, rifampin, dexamethasone, phenobarbital, phenytoin | Azole antifungals, erythromycin and cimetidine | CYP3A7 is functionally active in the foetus; approximately 30 to 75 % of adult levels of CYP3A4 |
Developmental Changes of Specific Cytochromes
Cytochrome P4501A2 (CYP1A2) accounts for much of the metabolism of caffeine [148] and theophylline [149]; methylxanthines are frequently used to treat neonatal apnoea and bradycardia. They are effective because they have prolonged action in neonates. CYP1A2 activity is nearly absent in the foetal liver and remains minimal in the neonate. This limits N-3- and N-7-demethylation of caffeine in the neonatal period that prolongs elimination in preterm and term neonates. Elimination is through the immature renal system, and consequent clearance is reduced. An adult clearance rate is achieved within the first postnatal year (Table 25.4) [150]. A similar pharmacokinetic pattern of reduced metabolism at birth occurs with theophylline (Table 25.5) in which CYP1A2 catalyses 3-demethylation and 8-hydroxylation [151]. Both drugs illustrate Michaelis–Menten metabolism after overdose in neonates, resulting in prolonged toxic effects [150, 151].
Table 25.4
Maturation of caffeine clearance
Age | Weight (kg) | CL per kilogram (L h−1 kg−1) | CL allometric (L h−1 70 kg−1) |
|---|---|---|---|
Premature neonate | 2 | 0.0041 | 0.118 |
Term neonate | 3.5 | 0.004 | 0.132 |
3 months | 6 | 0.091509 | 3.46 |
6 months | 7.5 | 0.119719 | 4.8 |
1 year | 10 | 0.126694 | 5.5 |
Adult | 70 | 0.057–0.085 | 3.6–5.9 |
Table 25.5
Maturation of theophylline clearance
Age | Weight (kg) | CL per kilogram (L h−1 kg−1) | CL allometric (L h−1 70 kg−1) | V (L kg-1) |
|---|---|---|---|---|
Neonate | 3 | 20 | 0.38 | 0.86 |
Infant (4 months) | 5 | 30–60 | 0.9–1.9 | |
Toddler (1 year) | 10 | 90 (SD 18) | 3.9 (SD 0.8) | 0.63 |
Child (2 years) | 12 | 76 (20) | 3.4 (0.9) | |
Child (7 years) | 22 | 62 (20) | 3.3 (1.0) | |
Adolescent (13 years) | 40 | 55 (18) | 3.3 (1.0) | |
Adult | 70 | 40 (12) | 2.8 (0.8) | 0.5 |
Other P450 enzymes that are reduced or absent in the foetus include CYP2D6 and CYP2C9 [144, 152, 153]. CYP2D6, which is involved in the metabolism of β blockers, antiarrhythmics, antidepressants, antipsychotics, tramadol and codeine, is absent in the foetal liver and is eventually expressed postnatally [154–156]. In contrast to the slow maturation of CYP1A2 and CYP2D6, CYP2C9, which is responsible for the metabolism of non-steroidal anti-inflammatory drugs (NSAIDs), warfarin and phenytoin, has minimal activity antenatally and then develops rapidly postnatally [154, 157].
CYP3A is the most important cytochrome involved in drug metabolism, because of the broad range of drugs that it metabolises and because it comprises the majority of adult human liver cytochrome P450 (see Table 25.3). CYP3A is detectable during embryogenesis as early as 17 weeks, primarily in the form of CYP3A7, and reaches 75 % of adult activity by 30 weeks’ gestation [158]. In vivo, CYP3A activity appears to be mature at birth; however, there is a poorly understood postnatal transition from the foetal CYP3A7 to the predominant adult isoform CYP3A4 [159]. This transition from CYP3A7 to 3A4 explains the similar clearance of methadone between neonates and adults (Fig. 25.16) [143]. Bupivacaine is metabolised by CYP3A4. This immature clearance of bupivacaine resulted in high plasma concentrations that caused seizures in neonates given epidural infusion at rates greater than that at which it was metabolised [160].
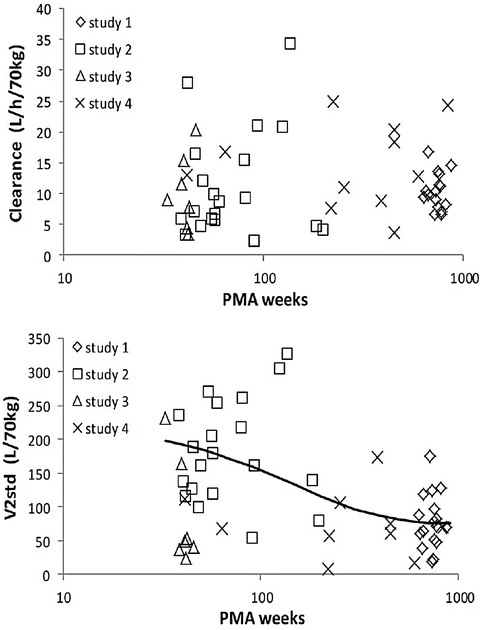
Fig. 25.16
The upper panel shows individual predicted methadone clearances, standardised to a 70 kg person, plotted against postmenstrual age. No relationship between age and standardised clearance was found. Individual predicted methadone peripheral compartment volume of distribution (V 2), standardised to a 70 kg person, is plotted against postmenstrual age in the lower panel. The solid line represents the nonlinear relation between V 2 and age (Reproduced from Ward RM, Drover DR, Hammer GB, Stemland CJ, Kern S, Tristani-Firouzi M, Lugo RA, Satterfield K, Anderson BJ. The pharmacokinetics of methadone and its metabolites in neonates, infants, and children. Pediatr Anesth 2014;24: 591–601 [143], with kind permission from John Wiley and Sons)
Phase II Reactions
The other major route of hepatic drug metabolism, designated phase II reactions, involves synthetic or conjugation reactions that increase the hydrophilicity of molecules to facilitate renal elimination. The phase II enzymes include glucuronosyltransferase, sulphotransferase, N-acetyltransferase, glutathione S-transferase and methyl transferase.
Most conjugation reactions have limited activity during foetal development. One of the most familiar synthetic reactions in young infants involves conjugation by uridine diphosphoglucuronosyltransferases (UGT). This enzyme system (also responsible for bilirubin) has numerous isoforms that all mature at different rates. Failure to appreciate UGT immaturity at birth resulted in high concentrations of chloramphenicol and consequent fatal circulatory collapse in neonates [161, 162].
Morphine , acetaminophen and dexmedetomidine all undergo glucuronidation . Morphine clearance [163] increases with weight and postmenstrual age. The maturation of glucuronosyltransferase enzymes varies among isoforms, but, in general, adult activity is reached by 6–12 months of age. Some of the confusion relating to maturation rates is attributable to the use of the per kilogram size model. The use of allometry with a maturation model has assisted understanding. The time courses of maturation of drug metabolism (morphine [45], acetaminophen [164], dexmedetomidine [165] and GFR [59]) are strikingly similar (Fig. 25.17) with 50 % of size-adjusted adult values being reached between 8 and 12 weeks (TM50) after full-term delivery. All three drugs are cleared predominantly by UGT that converts the parent compound into a water-soluble metabolite that is excreted by the kidneys. Glucuronidation is the major metabolic pathway of propofol metabolism, and this pathway is immature in neonates, although multiple cytochrome P450 isoenzymes, including CYP2B6, CYP2C9 or CYP2A6, also contribute to its metabolism and cause a faster maturation profile than expected from glucuronide conjugation alone [166]. A phase I reaction (CYP2D6) is the major enzyme system tramadol, and clearance through this pathway is faster than those associated with UGT maturation. Table 25.6 outlines some typical phase II processes.
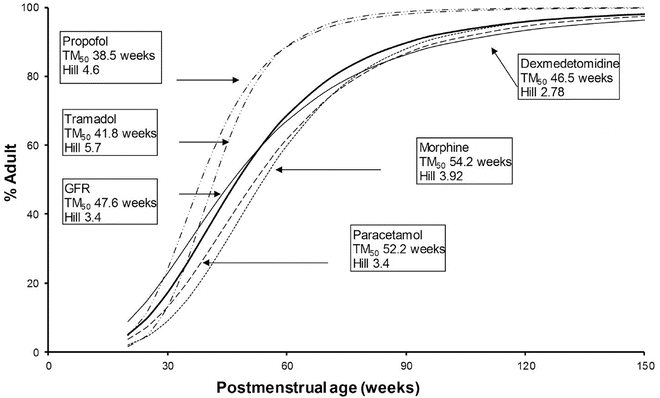
Fig. 25.17
Clearance maturation, expressed as a percentage of mature clearance, of drugs where glucuronide conjugation (paracetamol, morphine, dexmedetomidine) plays a major role. These profiles are closely aligned with glomerular filtration rate (GFR). In contrast, cytochrome P450 isoenzymes also contribute to propofol and tramadol metabolism and cause a faster maturation profile than expected from glucuronide conjugation alone. Maturation parameter estimates were taken from [45, 59, 156, 164, 165, 733]
Table 25.6
Developmental patterns for conjugation (phase II) reactions in the neonate
Enzymes | Selected substrates | Developmental patterns |
|---|---|---|
Uridine diphosphoglucuronyltransferase (UGT) | Chloramphenicol, morphine, hydromorphone, acetaminophen, valproic acid, lorazepam, dexmedetomidine, naloxone | Ontogeny is isoform specific. In general, adult activity is achieved by 6–18 months of age. Induced by cigarette smoke and phenobarbital |
Sulphotransferase | Bile acids, acetaminophen, cholesterol, polyethylene, glycols, chloramphenicol | Ontogeny seems to be more rapid than UGT; however, it is substrate specific |
N-acetyltransferase | Hydralazine, procainamide, clonazepam, caffeine, sulfamethoxazole | Some foetal activity present by 16 weeks. Adult activity present by 1–3 years of age |
Alterations in Biotransformation
Transition from the intrauterine to the extrauterine environment is associated with major changes in blood flow. There may also be an environmental trigger for the expression of some metabolic enzyme activities resulting in a slight increase in maturation rate above that predicted by PMA at birth (Fig. 25.18) [26, 166]. Many biotransformation reactions, especially those involving certain forms of cytochrome P450, are inducible before birth through maternal exposure to drugs, cigarette smoke or other inducing agents. Postnatally, biotransformation reactions may be induced through drug exposure and may be slowed by hypoxia/asphyxia, organ damage and/or illness. Postnatal changes in hepatic blood flow, protein binding and/or biliary function may also alter drug elimination.
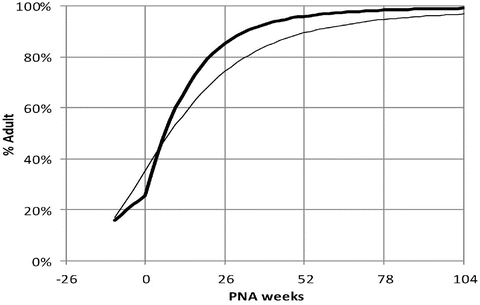
Fig. 25.18
Change in glomerular filtration rate associated with maturation and birth at 40 weeks PMA. The maturation profile determined using PMA is shown as a thin line. The effect of adding PNA is shown as a thick line. Maturation is slower than anticipated before birth when PNA is unaccounted for, and there is a slight increase of clearance maturation after birth (Reproduced from Anderson BJ, Holford NH. Tips and traps analyzing pediatric PK data. Pediatr Anesth 2011; 21: 222–37 [25] with kind permission from John Wiley and Sons)
Genotypic Variations in Drug Metabolism; CYP2D6
Single nucleotide changes or polymorphisms (SNPs) in the DNA sequence in CYP enzymes may decrease or increase metabolic activity for a specific drug substrate [167]. Variability in the clinical response to codeine prompted investigations into genetic variants or polymorphisms of CYP2D6, the enzyme that converts codeine to its active metabolite, morphine [168]. This enzyme is mapped to chromosome 22 at 22q13.1. Over 55 polymorphisms of CYP2D6 have been described with a frequency that exceeds 1 % of the population [169]. These include both functional and nonfunctional polymorphisms as well as gene duplication. The polymorphisms are numbered with *1 being the normal or wild allele (the * denotes an allele). The mutant alleles, *3, *4, *5, *6 and*9, for example, confer no CYP2D6 activity [169–171]. The latter polymorphisms account for more than 90 % of the poor metabolisers. Variants *2, *10 and 17 have modestly reduced activity and are referred as intermediate metabolisers. To further complicate the genetic pattern, multiple copies of the same genes [171] may be present in some individuals, resulting in bizarre phenotypes. The wide array of CYP2D6 polymorphisms of codeine may be summarised into three broad categories: poor metabolisers (negligible morphine produced [PM]), extensive metabolisers (normals [EM]) and ultra-extensive metabolisers (rapid and large amounts of morphine [UM]). Up to 10 % of whites and 30 % of Hong Kong Chinese are PM, rendering codeine an ineffective analgesic for these children. Alternately, 29 % of the Ethiopian and 1 % of Swedish, German and Chinese populations are UM. Children with the UM genotype who also have upregulated opioid receptors as a result of chronic intermittent nocturnal hypoxia may be particularly vulnerable to a mishap after a usual or subclinical dose of codeine [172].
Reduced metabolism through genetic polymorphisms leads to exaggerated effects when administered in conventional doses for many other drugs [167, 173]. Succinylcholine clearance through pseudocholinesterase is a well-known example [174]. Genotyping has become a routine part of the evaluation before treatment with methotrexate for detection of reduced activity of thiopurine methyltransferase that may be lethal with treatment with conventional dosages [175]. The drug irinotecan, used to treat cancer, has an active metabolite that is metabolised by a glucuronide (UGT1A1), a pathway similar to that involved in morphine clearance (UGT2B7). A variant allele UGT1A1*28 has been identified that is associated with severe neutropenia and diarrhoea. Genetic testing in patients to identify this allele (present in 10 % Caucasians) has been shown to be beneficial [176]. Specific SNPs of CYP2C9, CYP2C19, CYP2D6, CYP3A and uridine diphosphate-glucuronosyltransferase 1A1 (UGT1A1) account for a sufficient number of adverse pharmacologic outcomes to warrant future routine clinical testing [177].
Genotype may have little impact in the neonate when clearance is poor (Fig. 25.19) [178]. There is also evidence that possession of the genotype does not necessarily equate with phenotype. The clearance of tramadol by those with a low CYP2D6 genotype score was not reduced in all children studied (Fig. 25.20) [156].
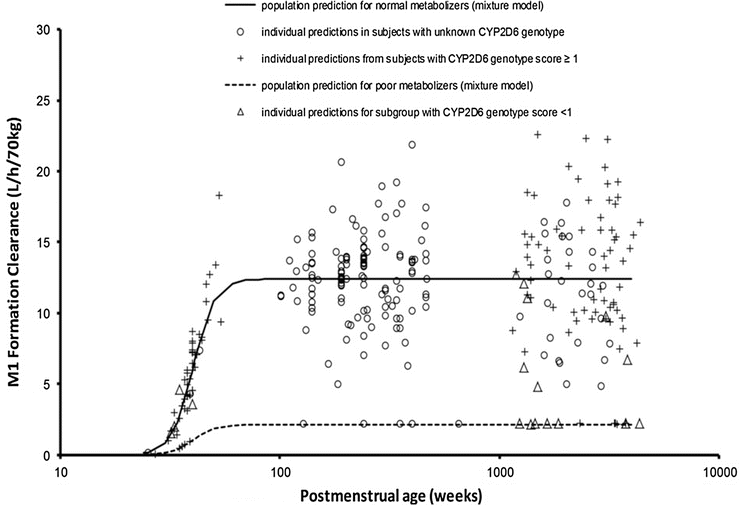
Fig. 25.19
Tramadol metabolite (M1) formation clearance (CYP2D6) increases with postmenstrual age. Rate of increase varies with genotype expression. Clearance is low in premature neonates, and genotype has minimal impact in early neonatal life (Adapted from Allegaert K, van den Anker JN, de Hoon JN, van Schaik RH, Debeer A, Tibboel D, Naulaers G, Anderson BJ. Covariates of tramadol disposition in the first months of life. Brit J Anaesth 2008;100:525–32 [178], with kind permission of Oxford University Press)
Fig. 25.20
Maturation of tramadol formation clearance (CLPM) to O-desmethyltramadol (M1 metabolite) labelled according to the availability of an individual CYP2D6 genotype activity score. There is a distinct group of patients who are slow metabolisers identified by the phenotype (dashed line). Not all subjects with a low genotype CYP2D6 activity (n = 20) are included in this slow metaboliser group. (From Allegaert K, Holford NHG, Anderson BJ, Holford S, Stuber F, Rochette A, Trocóniz IF, Beier H, de Hoon JN, Pedersen RS, Stamer U. Tramadol disposition throughout human life: a pooled pharmacokinetic study. Clin Parmacokinet 2015;54(2):167–78 [734], with kind permission from Springer)
Renal Excretion
Renal function in neonates and infants is less efficient than in adults due to the combination of incomplete glomerular development, low perfusion pressure and inadequate osmotic load to produce full countercurrent effects [179]. Preterm and term neonates have immature glomerular filtration and tubular function; both develop rapidly during the 6 months of life [59]. Glomerular filtration and tubular function are nearly mature by 20 weeks of postnatal age and fully mature by 2 years of age (Fig. 25.17) [59, 180]. Drugs eliminated primarily through the kidney, such as aminoglycoside antibiotics and milrinone, have reduced clearance in neonates and infants [181, 182].
In the presence of renal failure, one or two doses of drugs that are excreted via the kidneys often achieve and maintain prolonged therapeutic drug concentrations if there is no alternate pathway of excretion (e.g. caffeine or curare in the neonate). The PK and PD of the old muscle relaxant, curare, exemplify the complex interaction of increased V, smaller muscle mass and decreased rate of excretion due to immaturity of glomerular filtration. The initial dose of curare needed to achieve neuromuscular blockade is similar in infants and adults [183]. In infants, however, this blockade is achieved at reduced serum concentrations compared with older children or adults, corresponding to differences in muscle mass. A larger V (total body water) accounts for the equivalent dose for each kilogram of body weight, and the reduced glomerular function in infants compared with older children or adults accounts in part for the longer duration of action [183].
Pulmonary Elimination
The factors determining anaesthetic absorption (alveolar ventilation, FRC, cardiac output, tissue/blood solubility) also contribute to elimination. We might anticipate more rapid washout in neonates than adults for any given duration of anaesthesia because there is less distribution to fat and muscle content. The greater decrease in cardiac output induced by halothane in neonates might be expected to speed elimination, but brain perfusion will also be reduced and this slows recovery. Halothane in particular and to a far lesser extent isoflurane and sevoflurane undergo hepatic metabolism, but contribution is small compared to pulmonary elimination [184].
Extrahepatic Routes of Metabolic Clearance
Many drugs undergo metabolic clearance at extrahepatic sites. Remifentanil and atracurium are degraded by nonspecific esterases in tissues and erythrocytes. Thus, plasma clearances of atracurium and remifentanil are independent of organ function and, when normalised for body weight, are somewhat greater in neonates and infants than in older children [23, 185]. This relationship is demonstrated for remifentanil in Fig. 25.21. Clearance determines infusion rate at steady state, and it can be seen that remifentanil infusion rates required achieving a target respiratory rate mirror clearance. Succinylcholine also exhibits a monophasic decline in weight-normalised dose requirements from birth that is predictable by the 3/4 power size model [186–188].
Fig. 25.21
The effect of age on the dose of remifentanil tolerated during spontaneous ventilation under anaesthesia in children undergoing strabismus surgery. Superimposed on this plot is estimated remifentanil clearance determined using an allometric model. There is a mismatch between clearance and infusion rate for those individuals still in infancy. The higher infusion rates recorded in those infants can be attributed to greater suppression of respiratory drive in this age group than the older children during the study; a respiratory rate of ten breaths per minute in an infant is disproportionately slow compared to the same rate in a 7-year-old child, suggesting excessive dose (Reproduced from Anderson BJ. Paediatric models for adult target-controlled infusion pumps. Pediatr Anesth 2010;20:223–32 [735], with kind permission from John Wiley and Sons)
Paediatric Pharmacodynamic Considerations
Children’s responses to drugs have much in common with the responses in adults [189]. The perception that drug effects differ in children arises because the drugs have not been adequately studied in paediatric populations who have size- and maturation-related effects as well as different diseases. Neonates and infant, however, often have altered pharmacodynamics.
PD Differences in Neonates and Infants
The minimal alveolar concentration (MAC) fo r almost all anaesthetic vapours is less in neonates than in infancy, which is in turn greater than that observed in children and adults [128]. MAC of isoflurane in preterm neonates less than 32 weeks gestation was 1.28 %, and MAC in neonates 32–37 weeks gestation was 1.41 % [190]. This value rose to 1.87 % by 6 months before decreasing again over childhood [190]. The cause of these differences is uncertain and may relate to maturation changes in cerebral blood flow, gamma-aminobutyric acid (GABAA) receptor numbers or developmental shifts in the regulation of chloride transporters. Gamma-aminobutyric acid A receptor binding in human neonates is strikingly different from that in older children/adults [191].
Neonates have an increased sensitivity to the effects of neuromuscular blocking drugs [183]. The reason for this is unknown, but it is consistent with the observation that there is a threefold reduction in the release of acetylcholine from the infant rat phrenic nerve [192, 193]. The increased volume of distribution however means that a single NMBD dose is the same as the older child; reduced clearance prolongs duration.
The coagulation system [194, 195], including the fibrinolytic system at birth [196–199], is immature at birth. Consequently the target plasma concentration of antifibrinolytic drugs required to achieve similar effects as in adults is less in neonates. The concentration of epsilon aminocaproic acid (EACA) required to inhibit fibrinolysis in adult plasma in vitro was originally described to be 130 mg/L in 1962 [200]. This has been confirmed as the effective concentration in adults by using thromboelastography [201]. However, neonates require a lower concentration of EACA (50 mg/L) to inhibit fibrinolysis [202]. Dose of EACA requires adjustment in neonates because of both immaturity of antifibrinolytic clearance pathways and coagulation cascade.
Cardiac calcium stores in the endoplasmic reticulum are reduced in the neonatal heart because of immaturity. Exogenous calcium has greater impact on contractility in this age group than in older children or adults. Catecholamine release and response to vasoactive drugs vary with age. These PD differences are based in part upon developmental changes in myocardial structure, cardiac innervation and adrenergic receptor function. Conversely neonates may suffer cardiac arrest if given the calcium antagonist, verapamil [203]. There are some data to suggest greater sensitivity to warfarin in children, but the mechanism is not determined [204]. Amide local anaesthetic agents induce shorter block duration and require a larger weight-scaled dose to achieve similar dermatomal levels when given by subarachnoid block to infants. This may be due, in part, to myelination, spacing of nodes of Ranvier and length of nerve exposed as well as size factors. There is an age-dependent expression of intestinal motilin receptors and the modulation of gastric antral contractions in neonates. Prokinetic agents may not be useful in very preterm infants, partially useful in older preterm infants and useful in full-term infants. Similarly, bronchodilators in infants are ineffective because of the paucity of bronchial smooth muscle that can cause bronchospasm.
The sensitivity of human neonates to most of the sedatives, hypnotics and narcotics is clinically well known and may in part be related to increased brain permeability (immature blood–brain barrier or damage to the blood–brain barrier), particularly in the premature neonate, for some medications [93, 205–208]. Regional blood flow and receptor density differences within the brain also contribute. Decreased protein binding, as in the neonate, results in a greater proportion of unbound drug that is available for passive diffusion. This may contribute to bupivacaine’s propensity for seizures in neonates.
Measurement of PD Endpoints
Outcome measures are more difficult to assess in neonates and infants than in children or adults. Measurement techniques, disease and pathology differences, inhomogeneous groups, recruitment issues, ethical considerations and endpoint definition for establishing efficacy and safety confuse data interpretation [209].
Common effects measured include anaesthesia depth, pain and sedation and neuromuscular blockade. A common effect measure used to assess depth of anaesthesia is the electroencephalogram or a modification of detected EEG signals (spectral edge frequency, bispectral index, entropy). Physiological studies in adults and children indicate that EEG-derived anaesthesia depth monitors can provide an imprecise and drug-dependent measure of arousal. Although the outputs from these monitors do not closely represent any true physiological entity, they can be used as guides for anaesthesia and in so doing have improved outcomes in adults. In older children the physiology, anatomy and clinical observations indicate the performance of the monitors may be similar to that in adults. In infants their use cannot yet be supported in theory or in practice [210, 211]. During anaesthesia, the EEG in infants is fundamentally different from the EEG in older children; there remains a need for specific neonate-derived algorithms if EEG-derived anaesthesia depth monitors are to be used in neonates [212, 213].
The Children’s Hospital of Wisconsin Sedation Scale [214] has been used to investigate ketamine in the emergency department [70]. However, despite the use of such scales in procedural pain or sedation studies, few behavioural scales have been adequately validated in this setting [215, 216]. Interobserver variability can be high [217]. More objective measures for pain such as pupillometry may improve scoring [218], but these measures also contain thorns to their execution [219]. Most scores are validated for the acute, procedural setting and perform less for subacute or chronic pain or stress.
PKPD Parameter Variability
There is considerable variability in any measured plasma concentration when identical doses are given to individuals. Typical values for population PK parameter variability are 50 % for compliance with medication regimens, 30 % for absorption, 10 % for tissue distribution, 50 % for metabolic elimination and 20 % for renal elimination [220]. The contribution of PD variability due to distribution from the blood to the site of action will depend largely on changes in perfusion of target tissue (5–60 %). Receptor sensitivity variability (5–50 %) and efficacy variability (30 %) also exist. The observed response may not be a direct consequence of drug–receptor binding, but rather through intermediate physiological mechanisms. A typical value for this variability is 30 % [220].
Size and age are the two major contributors to variability of PK parameters (Fig. 25.22) [16]. Genetic differences in drug metabolism, receptor binding and intracellular coupling to effector mechanisms contribute variability to PK and PD. We know much less about morphine PD and response variability than we know about PK [2], but genetic influences contribute to morphine PD variability [221].
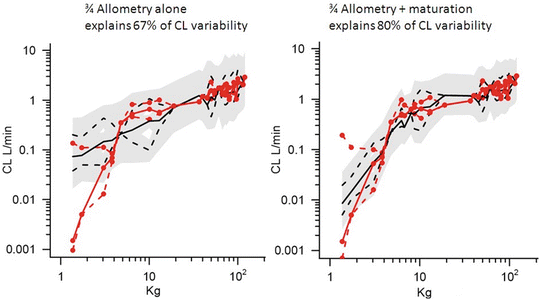
Fig. 25.22
Size and maturation explain much of propofol variability. Plots show median and 90 % intervals (solid and dashed lines). The 90 % prediction intervals for observations (lines with symbols) and predictions (lines) with 95 % confidence intervals for prediction percentiles (grey-shaded areas) are shown. (Reproduced from Anderson BJ, Holford NHG. Tips and traps analyzing paediatric PK data. Pediatr Anesth 2011;21:222–37 [26], with kind permission from John Wiley and Sons)
Genetic, physiological and cultural differences exist between men and women [222–224]. Hormones possibly have influence on the higher levels of pain and decreased pain threshold experienced by women because these differences are less significant after the age of 40 years. PK differences are not apparent before puberty [225], although PD differences may exist from an early age. Pain differences exist between people of different races and ethnicities. The contribution to these differences due to culture, geography [226, 227] and genetics remains to be teased apart. The incidence of adverse effects after morphine is greater in Latino than non-Latino children. Neither differences in morphine or metabolite concentrations nor the genetic polymorphisms examined explained these findings [228]. Caucasian children had a higher incidence of opioid-related adverse effects but less pain than African American children [229]. The latter had higher morphine clearance, and although UGT2B7 genetic variations (2161C > T and 802C > T) were not associated with observed racial differences in morphine’s clearance, the wild type of the UGT2B7 isozyme was more prevalent in African Americans [230].
The extremely anxious child may require a greater induction dose of propofol than less anxious children [231]. Increased circulating catecholamines may also contribute to perceived pain. Single nucleotide polymorphisms for the enzyme responsible for metabolising catecholamines (catechol-O-methyltransferase, COMT) have been described and distinct haplotypes categorised (low, average and high pain sensitivity). Haplotypes have also been associated with catecholamine synthesis (e.g. cofactor tetrahydrobiopterin (BH4) synthesis and metabolism) that is associated with chronic pain. BH4 blocking drugs may prove useful as a novel analgesic. In addition haplotypes for the B2 adrenergic receptor, based on eight single nucleotide polymorphisms, have been identified.
It is not surprising that inflammatory cytokines (interleukins, tumour necrosis factor) have impact on the pain response. The inflammatory response mediated after surgery has impact on pain [232]. Polymorphism in the interleukin-1 receptor antagonist gene is associated with serum interleukin-1 receptor antagonist concentrations and postoperative opioid consumption [233].
Morphine works through the μ-opioid receptor, a protein coded for by the OPRM1 gene on chromosome 6q24-q25. Polymorphisms of this gene (e.g. A118G) may increase this receptor’s affinity for morphine and its metabolite morphine 6-glucuronide [234] although clinical impact continues to be debated [235, 236] A number of other genetic variations may also influence the μ-opioid receptor. The melanocortin-1 receptor that serves a role in skin pigmentation may influence morphine 6-glucuronide effects as well as the k-opioid receptor in females [237]. Signal transmission from opioid receptors requires involvement of ion channels (K, Na, Ca), and polymorphisms of these channels have also been noted to have an influence on pain sensitivity. Mutations in voltage-gated transient receptor potential channels have been identified and may modulate the effects of analgesics [238]. Efflux transporters like the P-glycoproteins are also associated with polymorphisms and may affect transport into or out of the brain [239 ].
Adverse Effects
Neonates and young children may suffer permanent effects resulting from a stimulus applied at a sensitive point in development. For example, congenital hypothyroidism, if untreated, causes lifelong phenotypic changes. The incidence of vaginal carcinoma is high in children of mothers treated with stilboestrol during pregnancy [240]. There are concerns that neonatal exposure to some anaesthetic agents (e.g. ketamine, midazolam) may cause widespread neuronal apoptosis and long-term memory deficits [241, 242].
Anaesthesia, analgesia or sedation generally involves examination of immediate adverse effects such as PONV, hypotension or respiratory depression. A dose–response curve for intravenous morphine and vomiting was investigated in children having day-stay tonsillectomy. Doses above 0.1 mg kg−1 were associated with a greater than 50 % incidence of vomiting [243]. These data are similar to those in children undergoing inguinal herniorrhaphy [244], suggesting that lower doses of morphine are associated with a decreased incidence of emesis after day-stay surgery and encourage the use of alternative analgesic drugs.
Therapeutic use of drugs balances beneficial effects against adverse effects. Adverse effects, however, may be simply consequent upon a poor understanding of pharmacokinetics. Propofol infusion dose in neonates, if based on adult dose (mg/kg/h), will overdose and cause hypotension; propofol infusion dose in 1–2-year-olds (where clearance is increased expressed as mg/kg/h) may underdose and result in awareness. Morphine dose in the very young was traditionally limited by fears of respiratory compromise; postoperative arterial oxygen desaturation continues to be reported with sedative drugs in neonates [245]. These are a result of poor PK understanding. However there are also PD differences. Premature neonates are more prone to apnoea. Sympathetic–parasympathetic tone is immature in neonates, and the use of propofol in neonates has recently been associated with profound hypotension [246], questioning our understanding of the dose–effect relationships of this common drug [247]. Such information allows informed dosing.
Drug Interactions
Drug interactions can increase or decrease response mediated through either PK or PD routes. Physical drug interactions may occur even before absorption or delivery, reducing bioavailability (e.g. direct chemical combinations). Drug interactions may involve either PK interactions, PD interactions or even both. A commonly used combination in which PK interactions exist is that of midazolam and propofol. When given together, these drugs reduce the clearance of one another, resulting in a 25 % increase in propofol concentrations and a 27 % increase in midazolam concentrations [248, 249]. They also both act on gamma-aminobutyric acid (GABA) cerebral receptors contributing to PD interactions as well. Clinically this results in a 25 % reduction in required midazolam dose when given as bolus for short-term sedation with propofol in adults [248].
Pharmacokinetic Interactions
Interactions between co-administered drugs can affect all stages of PK processes (drug absorption, distribution, metabolism and elimination). A recent PK example is that of phenylephrine when administered orally with acetaminophen, typical of many over-the-counter preparations for upper respiratory tract infections [250]. This as yet unknown PK interaction results in nearly four times the normal maximum plasma concentration of phenylephrine, with changes to metabolism in the gut wall as one proposed mechanism [251].
PK interactions are often dealt with by including the effect of a second drug as a covariate on affected PK parameters such as those describing clearance (CL), volume of distribution (V) or bioavailability (F). The midazolam–propofol PK interaction has been investigated by adjusting midazolam CL and V using propofol plasma concentrations included in an exponential covariate model, i.e.
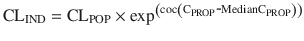
where CL is clearance from the central compartment, for the population (CLPOP) and the individual (CLIND). Here, the effect of plasma propofol concentration (CPROP) on the CL parameter is estimated (the parameter ‘cov’) and scaled to the population median CPROP (Median CPROP).
(25.22)
Phenobarbitone induces a number of other pathways responsible for drug clearances, e.g. CYP1A2, CYP2C9, CYP2C19, CYP3A4 and UDP-glucuronosyltransferase (UGT) [252]. Ketamine in humans is metabolised mainly by CYP3A4. The steep concentration–response curve described for ketamine [70] means that small changes in the plasma concentration attributed to increased clearance can have dramatic impact on the degree of sedation (Fig. 25.23) [253]. Another example relates to the administration of drugs that interfere with the cytochrome isoforms that metabolise midazolam (CYP3A4). Examples of such drugs/foods are grapefruit juice, erythromycin, calcium channel blockers and protease inhibitors. The net effect is to prolong the duration of action of midazolam.
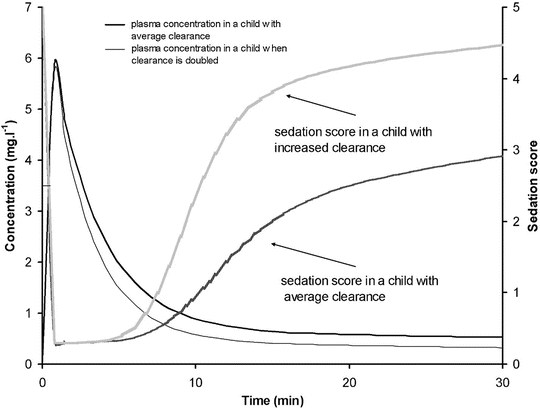
Fig. 25.23
Simulation of plasma concentration and sedation score in a 6-year-old, 20 kg child given ketamine 2 mg kg−1 when clearance is doubled. The sedation score is graded from 0 to 5 where a score of 2 indicates the child arouses slowly to consciousness, with sustained painful stimulus, and a score of 3 indicates the child arouses with moderate tactile or loud verbal stimulus (Reproduced from Sumpter A, Anderson BJ. Phenobarbital and some anesthesia implications. Pediatr Anesth 2011; 21: 995–7 [736], with kind permission from John Wiley and Sons)
Pharmacodynamic Models
Pharmacodynamics (PD) describes the concentration–effect relationship. PD models are often linked to PK models by describing movement of drug from the plasma to the effect site and its target (e.g. use of the equilibration half-time T 1/2 k eo), and interactions may even occur at that level. An increase in the T 1/2 k eo of d-tubocurarine with increasing inspired halothane concentrations has been demonstrated [254]. Halothane is a negative inotrope [255] and reduces skeletal muscle blood flow [256], so it seems reasonable to interpret changes in T 1/2 k eo as due to changes in blood flow.
Traditional methods of evaluating PD interactions include using isoboles, shifts in dose (or concentration) response curves (Fig. 25.24a) or interaction indices based on parameters of potency derived from separate monotherapy and combination therapy analyses. For example, inhalation anaesthetic agents can also prolong duration of neuromuscular block, and this affect is agent specific. Sevoflurane potentiated vecuronium more than halothane; when compared to balanced anaesthesia, the dose requirements of vecuronium were reduced by approximately 60 % and 40 %, respectively [257]. Such methods provide an estimation of the magnitude of effect for dose or concentration combinations, but they do not inform us on the time course of that effect or its associated variability.
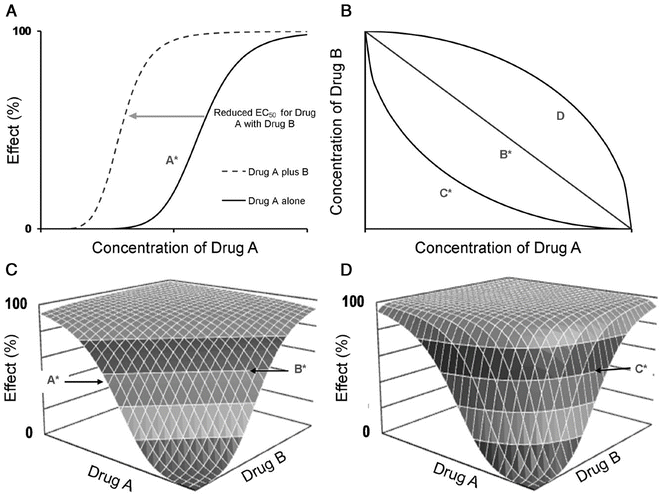
Fig. 25.24
Methods of investigating interactions. (a) Shift in response curve analyses involves plotting the concentration (or dose)–effect relationship for one drug alone and in the presence of steady-state concentrations of a second drug. (b) Isoboles are constructed using iso-effect lines with curves derived from observations assessed against the expected (or ‘additive’) response line (B*). Supra-additivity is depicted by curves bowing towards the plot origin (C*), while infra-additivity is shown with outward curves (D). Information from both methods is represented within response surfaces with isoboles displayed as horizontal planes and individual concentration–response curves as vertical slices (indicated by arrows on surfaces for A* single concentration–response curve drug A, B* additive isobole and C* supra-additive isobole). (c) Additive response surface for two drugs. (d) Synergistic response surface for two drugs, with synergy depicted through outward bowing of the surface (Reproduced from Hannam J, Anderson BJ. Pharmacodynamic interaction models in pediatric anesthesia. Pediatr Anesth 2015; 25:970–980 [737], with kind permission from John Wiley and Sons)
Drug effects can be described as a function of efficacy (e) and receptor occupation (i.e. the fraction of drug D bound to receptors R, [RD]), i.e.
![$$ E= f\ \left(\left[\mathrm{RD}\right]\cdotp e\right) $$](/wp-content/uploads/2017/07/A339434_1_En_25_Chapter_Equ23.gif)
PD interactions occur through various mechanisms that disrupt or alter this relationship. For example, competitive antagonists reduce receptor availability by competing for occupancy at the same receptor site. Drugs that elicit an effect are called agonists, while those that do not are called antagonists, so the occupancy of some receptors by the antagonists results in less effect. In general, competitive antagonists shift the effect–concentration curve to the right by altering the C 50. The E MAX equation can then be expressed as
![$$ \mathrm{Effect}={E}_0+\frac{E_{\mathrm{MAX}}\times {\mathrm{Ce}}^{\gamma}}{\left(\left({C_{50}}^{\gamma}\times \left[1+\frac{A}{{\mathrm{EA}}_{50}}\right]\right)+{\mathrm{Ce}}^{\gamma}\right)} $$](/wp-content/uploads/2017/07/A339434_1_En_25_Chapter_Equ24.gif)
where A and EA50 represent ligand A concentration and potency. Noncompetitive antagonists shift the observed maximum effect (E MAX) rather than the C 50.
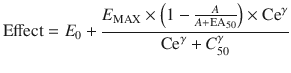
PD interactions are not restricted to same-site binding interactions; some proteins have multiple binding sites, and ligands binding at these sites can also alter the above relationship (i.e. through changes in protein conformation that lead to downstream changes or modulates agonist–receptor affinity).
(25.23)
(25.24)
(25.25)
Response Surface Models
A better way to investigate PD interactions is to use modelling and to take advantage of the benefits of population analyses. Models for monotherapy, derived using a population approach, can be combined and extended to incorporate PD interactions between two or more drugs.
The ‘response surface’ models are an extension of empirical, single drug models that can be used to describe, and predict, the combined effects between two or more drugs. The name refers to the surface of response that is visualised by plotting effect versus two drug concentrations (on x and y axes). Synergy (supra-additivity) is depicted by outward bowing of the surface on the horizontal plane, while infra-additivity is depicted by inward bowing of the surface (Fig. 25.24c, d). Horizontal lines within the response surface hold equivalent information to that given by isoboles (Fig. 25.24b). Surface parameters are estimated using data points pertaining to all areas of the concentration and effect range for both drugs simultaneously (e.g. as opposed to considering individual concentration pairs or effect levels in isolation, as is done with isobolographic analyses). The magnitude of the interaction is quantified by an interaction parameter. This is initially fixed at a value denoting the simplest scenario: no interaction or ‘additivity’. Resulting models can be used to characterise the type of interaction across the entire range of concentrations and effect levels and make predictions about effects for any ratio of the studied drugs.
Two equations are commonly used: those of Greco et al. [258] and Minto and Vuyk [259]. Greco equations have been used to describe additive effects for propofol, remifentanil and fentanyl on bispectral index response in children aged 1–16 years undergoing general surgery [65]. These authors reported C 50 estimates of propofol 5.20 μg/mL, remifentanil 24.1 ng/mL and fentanyl 8.6 ng/mL and suggested a propofol and remifentanil pair of 2.3 μg/mL and 4.3 ng/mL, respectively, to maintain haemodynamic parameters and sedation scores within ranges suitable for surgery. The Greco model has also been used to describe loss of response to various noxious stimuli under propofol–remifentanil anaesthesia [260]. Synergistic surfaces for sedation and response to laryngoscopy were reported.
Minto equations have been used to assess synergy for hypnosis between three commonly combined drugs for anaesthesia: propofol, midazolam and alfentanil [261]. Computer simulations based on interactions at the effect site predicted that the maximally synergistic three-drug combination (midazolam, propofol and alfentanil) tripled the duration of effect compared with propofol alone. Response surfaces can describe anaesthetic interactions, even those between agonists, partial agonists, competitive antagonists and inverse agonists [261].
Synergism between propofol and alfentanil has been demonstrated using response surface methodology. Remifentanil alone had no appreciable effect on response to shaking and shouting or response to laryngoscopy, while propofol could ablate both responses. Modest remifentanil concentrations dramatically reduced the concentrations of propofol required to ablate both responses [262]. When comparing the different combinations of midazolam, propofol and alfentanil, the responses varied markedly at each endpoint assessed and could not be predicted from the responses of the individual agents [263]. Similar response surface methodology has been taken for investigation of the combined administration of sevoflurane and alfentanil [264] and remifentanil and propofol [265] on ventilation control. These combinations have a strikingly synergistic effect on respiration, resulting in severe respiratory depression in adults. These synergistic associations can be extended to paediatric sedation techniques. It is little wonder that the use of three or more sedating medications compared with 1 or 2 medications was strongly associated with adverse outcomes [266].
The ability of propofol to ablate response to noxious stimuli has been studied in children aged between 3 and 10 years undergoing oesophagogastroduodenoscopy [267]. The C 50 for 50 % probability of no response was found to be reduced from a propofol concentration of 3.7 μg/mL to 2.8 μg/mL in the presence of 25 ng/kg/min remifentanil. The dose of remifentanil above this level did not result in large reductions in propofol requirements but did increase the risk of remifentanil-related respiratory depression.
‘Ketofol ’ is a mixture of ketamine and propofol (1:1) that is finding a niche for procedural sedation in the emergency room [268]. Stable haemodynamics, analgesia and good recovery are reported [269]. The additive interaction for anaesthesia induction in adults has been reported [270]. These data have been used to simulate effect in children [271]. An optimal ratio of racemic ketamine to propofol of 1:5 for 30 min anaesthesia and 1:6.7 for 90 min anaesthesia was suggested (Fig. 25.25) [271]. The ‘ideal mix’ for sedation will depend on the duration of sedation and the degree of analgesia required. The context-sensitive half-time of ketamine increases with infusion duration, resulting in delayed recovery [272].
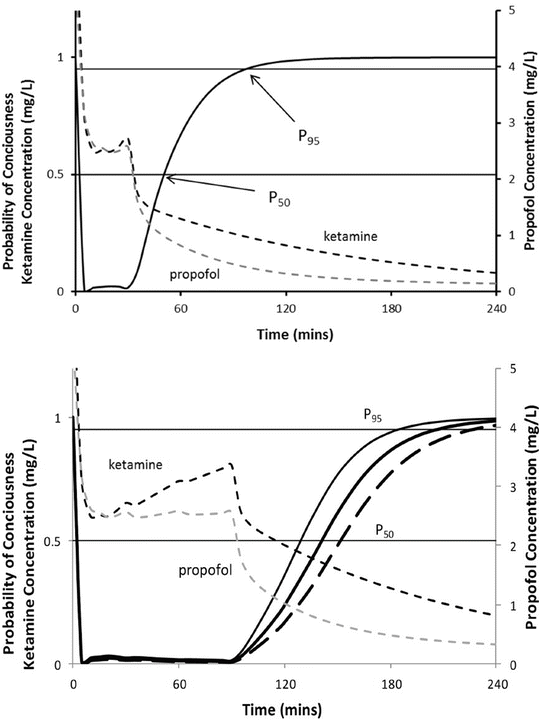
Fig. 25.25
The upper panel shows the probability of response during anaesthesia using a propofol/ketamine ratio of 5:1. The loading dose for induction of anaesthesia was 2.5 mg kg−1 propofol and 0.5 mg kg−1 of ketamine. Infusion rate was 67 % that suggested by McFarlan et al. [7]. The lower panel shows simulation results for a 90 min infusion. This panel also shows the probability of response as age increases from a 2-year-old (thin black line) to a 5-year-old (bold black line) and for a 10-year-old (bold black hash line) child (Reproduced from Coulter FL, Hannam JA, Anderson BJ. Ketofol dosing in anaesthesia. Pediatr Anesth 2014;24:806–12 [271], with kind permission from John Wiley and Sons)
Intravenous Anaesthetic Agents
Intravenous anaesthetics are a heterogeneous group of sedative–hypnotic drugs that produce unconsciousness speedily when injected intravenously. Prompt awakening after a single dose of these agents occurs predominantly by redistribution.
Barbiturates
Propofol
Propofol is an isopropylphenol supplied as a 1, 2 or 10 % aqueous solution containing soybean oil, glycerol and purified egg phosphatide to improve solubility. Its use for induction and maintenance of anaesthesia is associated with rapid recovery. Although the package insert for propofol cautions against its use in all patients with ‘egg allergy’, the evidence for this is not convincing [273, 274]. Propofol also suppresses laryngeal and pharyngeal reflexes, thereby facilitating tracheal intubation and the insertion of a laryngeal mask airway [275–277]. Emergence delirium rarely occurs after propofol anaesthesia in children. Propofol reduces the incidence of nausea and vomiting when used as an induction agent or when used for the maintenance of anaesthesia [278]. In view of these advantages, propofol has replaced thiopental as the induction agent of choice.
Pharmacokinetics
Propofol clearance matures rapidly in the 6 months of life (Fig. 25.17) The shorter distribution half-time and more rapid plasma clearance of propofol are responsible for the faster and more clear-headed recovery following a single dose of this agent compared with thiopental [279]. The rapid elimination of propofol (plasma clearance up to ten times faster than thiopental in some adult studies) reduces the potential for accumulation, making the drug suitable for maintenance of anaesthesia. Induction and maintenance doses of propofol are higher in children than in adults because the volume of the central compartment is 50 % larger and the plasma clearance (per kilogram) 25 % faster in children [280, 281]. Average induction doses (1.3 × ED50) in infants and children and adults are 4, 3 and 2 mg/kg, respectively [282, 283]. Clearance is limited by the hepatic blood flow and is consequently reduced in children in low cardiac output states.
Clearance (per kilogram) is increased in children; consequently a higher infusion (expressed as L/h/kg) dose is required to achieve the same target concentration as adults [7, 34]. These increased requirements in children can be attributed to size factors. There are a number of paediatric parameter sets that can be used for propofol infusion targeting a plasma concentration, e.g., Marsh et al. [12] and Gepts et al. [13], Kataria et al. [8], Short et al. [284], Rigby-Jones et al. [15], Schuttler and Ihmsen [55], Murat et al. [285], Saint-Maurice et al. [286], Coppens et al. [287] or Absalom et al. [14] (Table 25.1). Although parameter estimates are different for each author, most predict similar concentrations for the same infusion regimen (Fig. 25.3). Covariate influences such as severity of illness are unaccounted for; the volume in the central compartment, for example, is increased in children after cardiac surgery [15].
Pharmacodynamics
A propofol concentration of 2–3 mg/L is commonly sought for sedation, while 4–6 mg/L is used for anaesthesia. Both the loss and return of consciousness occur at similar target effect site propofol concentrations (2.0 SD 0.9 mg/L vs. 1.8 SD 0.7 mg/L) in adults [288], and a ‘wake-up’ concentration of 1.8 mg/L is described in children [289]. The relation between drug concentration and effect may be described by the Hill equation (Eq. (25.19)) [42]. Jeleazcov et al. [65] have described propofol pharmacodynamics in children 1–16 years using BIS where E 0 was estimated as 93.2, E max −83.4, EC50 5.2 mg L−1 and y 1.4. This relationship is very similar to that described in obese children [53]. The rate constant (k eo) describing for the effect compartment was 0.6 min−1 (T 1/2 k eo 1.15 min). Children possibly have a slightly lower sensitivity to propofol than adults (Fig. 25.26) [19], although this difference may be due to pharmacokinetic rather than pharmacodynamic factors [290]. The Kataria parameter set is known to underpredict concentration as age increases, consistent with allometric scaling. When this parameter set is used to estimate PD parameters, it appears that the older children require lower concentration to maintain anaesthesia [291]; this is a PK effect and not a PD effect [292].
Fig. 25.26
Propofol concentration and its relationship with bispectral index in children and adults (Data from Coppens et al. Anesthesiology 2011;115:83–93 and Chidambaran et al Pediatr Anesth 2015;25:911–23)
Decreased requirements in neonates are due to immature enzyme clearance systems. Propofol infusion rates for infants have been suggested [293]. Those regimens were determined by adapting an adult dosage scheme to the requirements of the younger population. Total number and time of administration of boluses and time to awakening were registered and used as criteria to adjust the dosage scheme. Predicted infusion rates are high (e.g. 24 mg kg−1 h−1 for the first 10 min in neonates) and should be used cautiously. Delayed awakening, hypotension and an increased incidence of bradycardia were reported in neonates and infants [293]. Propofol can cause profound hypotension in neonates, and pharmacokinetic–pharmacodynamic relationships in this age group remain elusive [247].
Adverse Effects
Pain on injection of propofol is a major problem occurring in up to 85 % of children [294]. It can be minimised by injecting into a large vein, injecting the solution slowly and administering at least 0.2 mg/kg of lidocaine immediately before, or with, propofol [295]. The vasodilator effects of propofol are greater than those of thiopental. Paediatric studies have consistently demonstrated a reduction in systolic and mean arterial pressures ranging from 5 to 30 % occurring in the first 5 min following injection of propofol [296–298]. Heart rate changes are variable in older children, but in one study, heart rate decreased significantly more in toddlers after propofol than after thiopental [296].
The use of propofol for prolonged sedation in paediatric intensive care units is associated with a rare syndrome comprising metabolic acidosis, heart failure, lipemia, rhabdomyolysis and death. The cause of this is unknown, but recent attention has focused on impairment of fatty acid oxidation by propofol as a possible cause [299, 300]. The fact that this propofol infusion syndrome is more common in children than adults may be a reflection of the higher dose requirement for propofol in children [301].
Thiopental
Thiopental is an analogue of pentobarbital, in which the oxygen attached at C 2 of the barbituric acid ring is replaced by sulphur. This substitution confers high lipid solubility, which in conjunction with a high cerebral blood flow results in rapid penetration of the brain and hypnosis. Elimination occurs by oxidation in the liver to an inactive metabolite which is excreted by the kidney.
In the neonate, plasma protein binding of thiopental is reduced, so that the fraction of unbound drug is almost twice that found in older children and adults [302, 303]. In addition, clearance at 26 weeks PMA was 0.015 L/min/70 kg and increased to 0.119 L/min/70 kg by 42 weeks PMA (approximately 40 % of adult clearance at term [304]); however, as recovery depends mainly on redistribution, the effect of an induction dose is not significantly prolonged (Fig. 25.27).
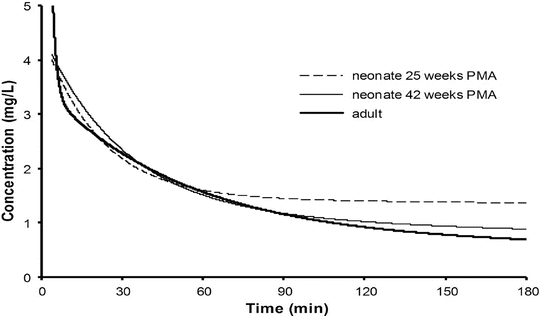
Fig. 25.27
Simulated time–concentration profiles after an intravenous thiopentone bolus of 3 mg kg−1. Predictions for a 25-week PMA and a 42-week PMA with neonate are shown alongside that of an adult (Reproduced from Larsson P, Anderson BJ, Norman E, Westrin P, Fellman V. Thiopentone elimination in newborn infants; exploring Michaelis Menten kinetics. Acta Anaesth Scand 2011; 55:444–51 [304], with kind permission from John Wiley and Sons)
Children aged 13–68 months given rectal thiopental (44 mg/kg) 45 min prior to surgery were either asleep or adequately sedated with plasma concentrations above 2.8 mg/L [305]. The ED50 sleep dose of intravenous thiopental varies with age [306, 307]. Doses of about 1.3 × ED50 of thiopental are required to produce rapid, reliable induction of anaesthesia in all age groups; thus, healthy neonates require about 4–5 mg/kg, infants 7–8 mg/kg and children 5–6 mg/kg of thiopental for induction. The reduced requirement for thiopental in neonates compared with infants age 1–6 months may be explained by decreased plasma protein binding [302], greater penetration of the neonatal brain [206] or increased responsiveness of neonatal receptors [308]. The increased requirements for thiopental in infants and children compared with adults (average adult dose 4 mg/kg) remain unknown. A pharmacodynamic explanation attributable to increased cerebral GABAA receptor numbers or maturational differences in relative organ mass and regional blood flow may contribute. Blood flow, relative to cardiac output, to kidney and brain increases, while that to the liver decreases in early life. Cerebral and hepatic masses as a proportion of body weight are much higher in the young child than in the adult.
A few seconds of apnoea followed by a period of respiratory depression is common after an induction dose of thiopental. The hypotensive response in neonates given thiopental appears not as dramatic as that associated with propofol, although it still may occur with reversion to foetal circulation. Thiopental has little direct effect on vascular smooth muscle tone, although myocardial depression may occur with induction [296, 309] related to the dose given and the rate of injection. Other reported adverse effects include hiccups, coughing and laryngospasm. Extravasation of thiopental or intra-arterial injection can cause tissue injury, probably due to its extreme alkalinity.
Thiopental has also been used as a continuous infusion (2–4 mg/kg/h) to control intracranial hypertension. The elimination of thiopental after a continuous infusion may be markedly prolonged compared with that after a single bolus (11.7 vs. 6.1 h) [310]. These findings may in part be attributed to the underlying illness, intercurrent drug treatment and zero-order kinetics at higher concentrations (Michaelis constant 28.3 mg/L) [304, 311].
Ketamine
Ketamine is a derivative of phencyclidine that similarly antagonises the N-methyl-d-aspartate (NMDA) receptor . Its action is related to central dissociation of the cerebral cortex, and it also causes cerebral excitation. Processed EEG monitoring devices do not work with ketamine sedation/anaesthesia [312].
Pharmacodynamics
Ketamine is available as a mixture of two enantiomers; the S(+)-enantiomer has four times the potency of the R(−)-enantiomer. S(+)-ketamine has approximately twice the potency of the racemate [313]. The metabolite norketamine has a potency that is one third that of its parent. Plasma concentrations associated with anaesthesia are approximately 3 μmcg/mL [272], hypnosis and amnesia during surgery are 0.8–4 cmg/mL and awakening usually occurs at concentrations less than 0.5 mcg/mL. Pain thresholds are increased at 0.1 mg/mL [314]. The concentration–response curve for ketamine sedation is steep [70, 315]. This means that small serum concentration changes will have dramatic effect on the degree of sedation observed (Fig. 25.28) [70]. Ketamine is very lipid soluble with rapid distribution, and the onset of anaesthesia after IV ketamine is approximately 30 s. The T 1/2 k eo was estimated ar 11 s [70].
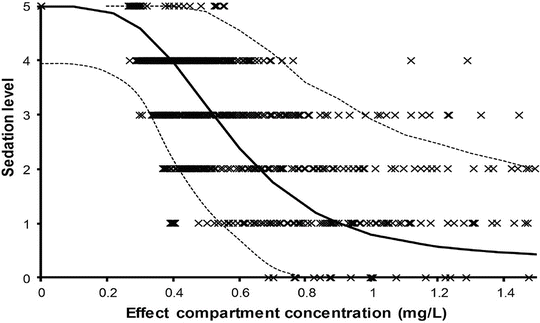
Fig. 25.28
The relationship between effect compartment concentration and level of sedation. The EC50 was 0.562 mg/L. Categorical data are shown as crosses. The dotted lines demonstrate 90 % confidence intervals (Reproduced from Herd D, Anderson BJ, Keene NA, Holford NHG. Investigating the pharmacodynamics of ketamine in children. Pediatr Anesth 2008;18:36–42 [70], with kind permission from John Wiley and Sons)
Pharmacokinetics
Children require greater doses of ketamine (mg/kg) than adults because of greater clearance (L/h/kg); however, there is considerable patient-to-patient variability [316, 317]. Ketamine undergoes N-demethylation to norketamine; metabolised mainly by CYP3A4, although CYP2C9, CYP2B6 also has a role. Elimination of racemic ketamine is complicated by the R(−)-ketamine enantiomer, which inhibits the elimination of the S(+)-ketamine enantiomer [318]. Clearance in children is similar to adult rates (80 L/h/70 kg, i.e. liver blood flow) within the first 6 months of life, when corrected for size using allometric models [84]. Clearance in the neonate is reduced (26 L/h/70 kg) [319–321], while V ss is increased in neonates (3.46 L/kg at birth, 1.18 L/kg at 4 years, 0.75 L/kg at adulthood [319]). This larger V ss in neonates contributes to the observation that neonates require a fourfold greater dose than 6-year-old children to prevent gross motor movement [322]. The α-elimination half-life was 11 min and a β-elimination half-life was 2.5–3.0 h [323, 324]. S(+) ketamine clearance was found to be 35.8 mL kg−1 min−1 with 5 and 95 % confidence limits of 11.5 and 111.1 mL/kg/min, respectively, in adults [325]. Context-sensitive half-time increases dramatically with use between 1 and 2 h infusion causing delayed recovery (Fig. 25.29).
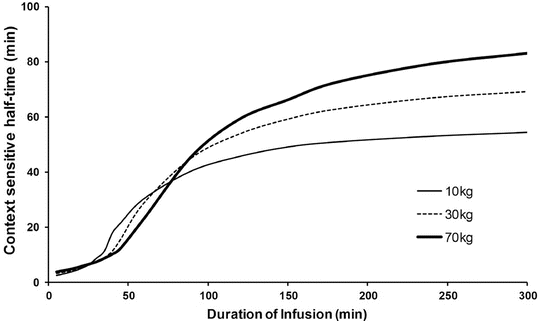
Fig. 25.29
Ketamine context-sensitive half-time following infusion at 3 mg/kg/h. The context-sensitive half-time in children was shorter than in adults after 1.5 h (Reproduced from Dallimore D, Anderson BJ, Short T, Herd DW. Ketamine anaesthesia in children—exploring infusion regimens. Pediatr Anesth 2008;18:708–14 [272], with kind permission from John Wiley and Sons)
Adverse Effects
The most common adverse reaction to ketamine anaesthesia is postoperative vomiting, which occurs in 33 % of children after doses used for anaesthesia [329]. Intraoperative and postoperative dreaming and hallucinations occur more commonly in older than in younger children [329]. The incidence of these latter adverse effects may be reduced when ketamine is supplemented with a benzodiazepine.
Atropine or another antisialagogue is commonly used to diminish the production of copious secretions that occur with ketamine. If an antisialagogue is not administered, there is a greater risk for laryngospasm [330], although guidelines for emergency departments suggest that supplementation with atropine or a benzodiazepine may not be necessary with lower doses [331–333]. Even small doses have the potential for apnoea or airway obstruction, particularly when combined with other sedating medications [334–336].
Ketamine increases heart rate, cardiac index and systemic blood pressure in adults [337]. In children, there is apparently no effect on pulmonary artery pressure provided that ventilation is controlled [338, 339]. Ketamine sedation has been shown to maintain peripheral vascular resistance, thus affecting intracardiac shunting less than propofol in children sedated for cardiac catheterisation [340]. Ketamine has negative inotropic effects in those who depend on vasopressors [341].
Ketamine may produce increases in intracranial pressure (ICP) as a result of cerebral vasodilation and may be contraindicated in children with intracranial hypertension. This concern regarding ICP has been challenged [342, 343]; control of respiration and consequent CO2 ameliorate changes in ICP [344, 345]. A 30 % increase in IOP has also been noted; thus, ketamine may be potentially dangerous in the presence of a corneal laceration [346].
Animal studies have correlated ketamine treatment with increased neuronal apoptosis during rapid synaptogenesis after birth [347–349]. It is unclear whether these data can be extrapolated from animals to developing humans [350]. Similar observations in rodents have been made with isoflurane, nitrous oxide, benzodiazepines and other medications commonly used to provide sedation/analgesia and anaesthesia to infants.
Etomidate
Etomidate is a steroid-based hypnotic induction agent. It is metabolised principally by hepatic esterases. Concentrations associated with anaesthesia are 300–500 mcg/L. As with most induction agents, offset of effect is by redistribution; clearance is approximately 1000 mL/min/70 kg in children and adults.
Etomidate pharmacokinetics have been studied in children with a median age of 4 years (range 0.53–13.21 years) and weight 15.7 kg (7.5–52 kg). A three-compartment model found the most significant covariate was age, with increasing age having reduced size-adjusted CL, V 1 and V 3. The estimates of PK parameter (standardised to 70 kg adult) for a typical 4-year-old children were CL 1.50 L/min/70 kg, Q 2 1.95 L/min/70 kg, Q 3 1.23 L/min/70 kg, V 1 9.51 L/70 kg, V 2 11.0 L/70 kg and V 3 79.2 L/70 kg [351]. Similar to propofol, younger children require a larger etomidate bolus dose than older children to achieve equivalent plasma concentrations [351, 352].
Etomidate clearance is reduced in neonates and infants (postnatal age 0.3–11.7 months) with congenital heart disease. A two-compartment model with allometric scaling to a 70 kg adult revealed a CL 0.624 L/min/70 kg and Q 0.44 L/min/70 kg; central (V 1) and peripheral distribution volumes (V 2) were 9.47 L/70 kg and 22.8 L/70 kg, respectively. Interindividual variability was high (between 94 and 142 % for all parameters) attributable to maturation over the age span studied [353].
Etomidate is painful when administered intravenously. However, concerns regarding the risks of anaphylactoid reactions and suppression of adrenal function have resulted in most anaesthesiologists avoiding this induction agent in routine cases [354]. Novel etomidate derivatives without this adverse effect are under investigation [355]. Etomidate is very useful in children with head injury and those with an unstable cardiovascular status such as children with a cardiomyopathy because of the virtual absence of adverse effects on the haemodynamics or cardiac function [356, 357].
Opioids
Opioid agonists produce analgesia and respiratory depression by combining with μ- and κ-opioid receptors. The introduction of newer opioid drugs with short context-sensitive half-times has rekindled interest in the use of opioids to avoid the major cardiovascular effects of volatile agents.
Morphine
Brain uptake after intravenous dosing is slow due to poor lipid solubility and consequent slow passage across the blood–brain barrier. An equilibration half-time (T 1/2 k eo) of 16–23 min is reported for analgesia in adults [358–360]. Although a morphine concentration–response curve for analgesia has not been described for children [221], morphine may display similar pharmacodynamics for respiratory depression and analgesia [361]. An T 1/2 k eo estimate for the morphine respiratory depressant effect was 16 min in a child [362], similar to that reported for analgesia. The EC50 for morphine’s respiratory depressant effect of 10–18 ng/mL [360–363] is consistent with clinical observations for both analgesic concentrations (10–20 ng/mL) [364–366] and respiratory depression (hypercapnia in 46 % children with concentration >15 ng/mL) [92].
Empiric studies have taught us that a concentration range (10–20 mcg/L) has analgesic effect [364] without associated adverse effects such as the respiratory depression observed with higher concentrations [92] or postoperative nausea and vomiting reported with higher doses [243]. Fears of adverse effects, particularly respiratory depression, dictate that morphine dose is titrated to gain satisfactory analgesia [367, 368]. A regime such as a loading dose of 50 mcg/kg followed by 25 mcg/kg at 5 min intervals to control pain is a satisfactory method [369]. A smaller dose of 20 mcg/kg is used in neonates. The drug remains remarkably safe when used as an infusion in hospital practice. The overall incidence of serious harm was only 1:10 000 with opioid infusion techniques, and those predisposed to harm can be identified (e.g. young infants, those with neurodevelopmental, respiratory or cardiac comorbidities) [370, 371].
Infusion regimens are based on clearance. Conjugation with glucuronide (UGT2B7) produces both active (morphine-6-glucuronide) and inactive (morphine-3-glucuronide) metabolites, which are excreted by the kidneys [372]. Clearance increases from 3.2 L/h/70 kg at 24 weeks PMA to 19 L/h/70 kg at term, reaching adult values (80 L/h/70 kg) at 6–12 months (Fig. 25.17) [163, 372]. Oral bioavailability is approximately 35 %.
Infusion regimens that achieve a plasma concentration of 10 mcg/L can be predicted from clearance (Fig. 25.30). Morphine exhibits perfusion-limited clearance, and positive pressure ventilation, by reducing hepatic blood flow, also reduces clearance [163]. While hepatic failure may be an obvious cause for reduced clearance, it is little appreciated that renal failure may also reduce clearance by approximately 30 %. The kidney is also a major contributor to morphine–glucuronide conjugation [373–376]. Reduced morphine clearance has also been described in children with cancer [377, 378], following cardiac surgery [366], and in critically ill neonates requiring extracorporeal membrane oxygenation (ECMO) [44, 379], although contributions from hepatic failure, renal compromise or positive pressure ventilation towards these reduced clearances were not acknowledged.
Fig. 25.30
Age-based infusion dosing for morphine with a target average steady-state concentration of 10 mcg/L in children not receiving positive pressure ventilation. The dotted line is the predictions based on age and typical weight for age. The solid line is the suggested practical infusion rate dose in mcg/kg/h at different postnatal ages. PNA years = (PMA weeks 40)/52. The upper panel shows dose related to postnatal age. The lower panel shows dose related to PMA (Reproduced from Anderson BJ, Holford NHG. Understanding dosing: children are small adults, neonates are immature children. Arch Dis Child 2013; 98: 737–44 [2], with kind permission from the BMJ Publishing Group)
Around 60 % of morphine is converted to morphine-3-glucuronide (M3G) and a further 6–10 % to morphine-6-glucuronide (M6G). M6G also has both analgesic and respiratory depressive effects. Animal data suggest that morphine and M6G act via distinct μ-receptor pathways and that an additive relationship exists between morphine and M6G for analgesic effects [380, 381]. The M6G T 1/2 k eo for respiratory depression is within the 4–8 h range [362], similar to reported for delayed M6G analgesic effect [360]. The hypercapnoeic ventilator responses for both parent and metabolite are similar (EC50 10–18 ng mL−1) [360–363]. The response curve steepness (represented by the Hill parameter of 2.4) for morphine and M6G was also similar in a case study [362]; this has also been noted by others examining the hypercapnoeic ventilator [360] and pupillary responses to morphine and M6G [382]. Respiratory depression in a child with renal failure who was given morphine postoperatively was best described by additive morphine and M6G respiratory effects (Fig. 25.31).
Fig. 25.31
Observed and predicted respiratory rate for a 12-year-old male child with end-stage renal failure given intravenous morphine via patient-controlled analgesia pump for postoperative pain. Predicted respiratory rates are for morphine only, M6G only and combined morphine–M6G effect. Observed partial pressure of CO2 (κPa) is also given. Dialysis began at 45 h postoperatively (indicated by arrow) and continued for 3 h. (Reproduced from Hannam J, Anderson BJ. Contribution of morphine and morphine 6-glucuronide to respiratory depression in a child. Anaesth and Intens Care 2012;40:867–70 [362], with the kind permission of the Australian Society of Anaesthetists)
Fentanyl
Fentanyl is a synthetic opioid with lipid solubility about 600 times greater than that of morphine. High lipid solubility confers increased potency, rapid onset (T 1/2 k eo 6.6 min in adults) and short duration of action. Fentanyl is a potent μ-receptor agonist with potency 100 times greater than that of morphine. A plasma concentration of 15–30 mcg/L is required to provide total intravenous anaesthesia (TIVA) in adults, whereas the EC50, based on EEG evidence, is 10 mcg/L [383, 384]. After a dose of 1–2 μg/kg, the clinical effects of fentanyl are terminated by redistribution, and its duration of action is limited to 20–30 min. However, after repeated doses or a continuous infusion, progressive saturation of peripheral compartments will result in prolonged duration of action.
Fentanyl is metabolised by oxidative N-dealkylation (CYP3A4) into norfentanyl and hydroxylated fentanyl. Clearance in preterm neonates is markedly reduced (T 1/2 β is 17.7 h) contributing to prolonged respiratory depression in that population. The clearance of fentanyl is reduced to 70–80 % of adult values in term neonates and, when standardised using allometry, reaches adult values (approx. 50 L/h/70 kg) within the first 2 weeks of life [84]. Clearance of fentanyl in older infants (>3 months of age) and children is greater than in adults when expressed as per kilogram (30.6 mL/kg/min vs. 17.9 mL/kg/min, respectively) resulting in a reduced elimination half-time (T 1/2 β 68 min vs. 121 min, respectively). These age-related changes follow the expected pattern portrayed in Fig. 25.10. Fentanyl clearance may be impaired with decreased hepatic blood flow (e.g. from increased intra-abdominal pressure in neonatal omphalocele repair); a maldistribution of blood away from regions of concentrated cytochrome enzyme activity in the liver may also play a role [385]. Fentanyl’s volume of distribution at steady state (V dss) is ~5.9 L/kg in term neonates and decreases with age to 4.5 L/kg during infancy, 3.1 L/kg during childhood and 1.6 L/kg in adults [78].
Infants with cyanotic heart disease had reduced V ss and greater plasma concentrations of fentanyl with infusion therapy [386]. These greater plasma concentrations resulted from a reduced clearance (34 L/h/70 kg) that was attributed to haemodynamic disturbance and consequent reduced hepatic blood flow [387]. Hypothermia has also been shown to reduce fentanyl clearance [388].
The infusion rates of fentanyl that are required to achieve a similar level of sedation/analgesia in critically ill children may vary as much as tenfold [389]. This variability in PK and PD strongly reinforces the need to titrate the dose to effect and to be prepared to provide postoperative ventilation support as needed. The context-sensitive half-time (CSHT) after a 1 h infusion of fentanyl is ~20 min, which increases to 270 min after an 8 h infusion in adults [27]. While the CSHT is reduced in children [390], there are no data in neonates. Children who receive a chronic infusion of fentanyl are at risk of rapidly developing tolerance to the opioid. On discontinuance of the infusion, these children may demonstrate signs of withdrawal. All long-term infusions should be tapered slowly over days rather than abruptly discontinuing the infusion [391, 392].
Chest wall and glottic rigidity have been reported after IV administration of all opioids, although most commonly after fentanyl [393, 394] One other concern is the rare association of increased vagal tone with bolus administration; bradycardia may have profound effects on the cardiac output of neonates. Additionally, fentanyl markedly depresses the baroreceptor reflex control of heart rate in neonates [395]. It is for these reasons that the combination of pancuronium and fentanyl became popular.
Remifentanil
Remifentanil is an ultrashort-acting synthetic opioid. Because of its ester linkage, it is susceptible to hydrolysis by nonspecific blood and tissue esterases. Its major metabolite is a carbolic acid compound with less than 0.3 % of the activity of the parent compound. Unlike other opioids, the duration of effect of remifentanil does not increase with increasing dose or duration of infusion, because its volume of distribution is small and its clearance is fast.
The target concentration may vary depending on the magnitude of desired effect. A remifentanil target of 2–3 mcg/L is adequate for laryngoscopy and 6–8 mcg/L for laparotomy, and 10–12 mcg/L might be sought to ablate the stress response associated with cardiac surgery [396]. Analgesic concentrations are 0.2–0.4 μg/L. Onset is rapid with a T 1/2 k eo is 1.16 min in adults [20]. Remifentanil clearance can be described in all age groups by simple application of an allometric model [23]. A standardised clearance of 2790 mL/min/70 kg is similar to that reported by others in children and adults. The smaller the child, the greater the clearance when expressed as mL/min/kg (Fig. 25.21). Clearance decreases with increasing age, with rates of 90 mL/kg/min in infants <2 years of age, 60 mL/kg/min in children 2–12 years of age and 40 mL/kg/min in adults. The steady-state volume of distribution was greatest in infants <2 months age (452 mL/kg) and decreased to 308 mL/kg in children 2 months–2 years and to 240 mL/kg in children >2 years of age [22].
Although covariate effects such as cardiac surgery appear to have a muted effect on PK, cardiopulmonary bypass (CPB) does have an impact. Remifentanil dosage adjustments are required during and after CPB due to marked changes in its volume of distribution [397]. Other PK changes during CPB are consistent with adult data in which a decreased metabolism occurred with a reduced temperature [398] and with reports of greater clearance after CPB (increased metabolism) compared with during CPB [24].
Respiratory depression is concentration dependent [399, 400]. Muscle rigidity remains a concern with bolus doses above 3 mcg/kg used for intubation in neonates [401]. The initial loading dose of remifentanil may cause hypotension and bradycardia [402] prompting some to target the plasma rather than effect site concentration when initiating infusion. This hypotensive response has been quantified in children undergoing cranioplasty surgery. A steady-state remifentanil concentration of 14 mcg/L would typically achieve a 30 % decrease in mean arterial blood pressure (Fig. 25.32). This concentration is twice that required for laparotomy, but is easily achieved with a bolus injection. The T 1/2 k eo of 0.86 min for this haemodynamic effect [66] is less than remifentanil-induced spectral edge changes described in adults (T 1/2 k eo = 1.34 min) [20, 403].
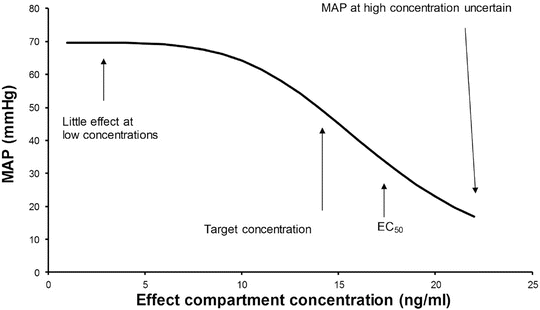
Fig. 25.32
The relationship between remifentanil concentration and mean arterial blood pressure (MAP) for a typical individual. A steady-state remifentanil concentration of 14 mcg/L would typically achieve a 30 % decrease in mean arterial blood pressure (Reproduced from Anderson BJ, Holford NHG. Leaving No Stone Unturned, Or Extracting Blood from Stone? Pediatr Anesth 2010;20:1–6 [403], with kind permission from John Wiley and Sons)
Adult remifentanil PK parameters [20] continue to be used in TCI devices for all ages, despite an increasing knowledge about this drug in children [21]. There is an element of safety with this approach because both volume of distribution [22] and clearance (expressed as mL min−1 kg−1) [23] decrease with age throughout childhood and because the elimination half-time is small with a constant context-sensitive half-time. The larger volume of distribution results in lower peak concentrations after bolus; the higher clearance in children results in lower plasma concentration when infused at adult rates expressed as mg min−1 kg−1. Owing to these enhanced clearance rates, smaller (younger) children will require higher remifentanil infusion rates than larger (older) children and adults to achieve equivalent blood concentrations.
The usual infusion dose of remifentanil is 0.1–0.5 μg/kg/min. The short elimination half-time frequently makes a loading dose unnecessary and facilitates control of the infusion. In a multicenter trial in full-term infants aged less than 2 months, remifentanil provided stable haemodynamic conditions with no new onset of postoperative apnoea [404]. Analgesic alternatives should be available for when the short-duration analgesic effect from remifentanil has dissipated. Reports of a rapid development of μ-receptor tolerance with remifentanil use are conflicting. Activity at δ-opioid receptors may contribute [405].
Alfentanil
Alfentanil (Alfenta) is a fentanyl analogue with reduced lipid solubility and smaller V compared with fentanyl [406, 407]. It has a rapid onset (T 1/2 k eo 0.9 min in adults), a brief duration of action and one fourth the potency of fentanyl. A target plasma concentration of 400 ng/mL is used in anaesthesia. Metabolism is through oxidative N-dealkylation by CYP3A4 and O-dealkylation and then conjugation to end products that are excreted via the kidney [408].
Clearance in neonates (20–60 mL/min/70 kg) is one tenth that in adults (250–500 mL/min/70 kg) [74] with rapid maturation. In preterm neonates, the half-life is as long as 6–9 h [409, 410]. The V in children and adults is similar, but increased in preterm neonates (1.0 SD 0.39 vs. 0.48 SD 0.19 L/kg). Clearance is greater in children expressed as per kilogram (11.1 SD 3.9 mL/kg/min vs. 5.9 SD 1.6 mL/kg/min). As a result, the elimination half-life in children is less than adults (63 SD 24 vs. 95 SD 20 min) [411–414]. Clinical effects are prolonged in those with reduced hepatic blood flow (e.g. increased intra-abdominal pressure, vasopressor use, some forms of congenital heart disease) [415, 416]. Because less alfentanil is bound to α1-acid glycoprotein in preterm infants (65 %) than in term infants (79 %), an increased fraction of alfentanil is available for biologic effect in the former [417].
The pharmacokinetics and pharmacodynamics of alfentanil have proven useful for the rapid control of analgesia and awakening from total intravenous anaesthesia [418, 419]. Alfentanil should be used with caution without neuromuscular blocking drugs in neonates because of the frequency of rigidity [420, 421].
Sufentanil
Sufentanil is a potent synthetic opioid that is similar to fentanyl and alfentanil. Sufentanil is five to ten times more potent than fentanyl, with a T 1/2 k eo of 6.2 min in adults [422]. A concentration of 5–10 ng/mL is required for TIVA and 0.2–0.4 ng/mL for analgesia. Pharmacodynamic differences are suggested in neonates. The plasma concentration of sufentanil at the time of additional anaesthetic supplementation to suppress haemodynamic responses to surgical stimulation was 2.51 mcg/L in neonates, significantly greater than the concentrations of 1.58, 1.53 and 1.56 mcg/L observed in infants, children and adolescents, respectively [423].
The volume of distribution at steady state (V ss) was 4.15 SD 1.0 L/kg in neonates, greater than the values of 2.73 SD 0.5 and 2.75 SD 0.5 L/kg observed in children and adolescents, respectively [423, 424]. Elimination of sufentanil has been suggested by O-demethylation and N-dealkylation in animal studies. Like fentanyl and alfentanil, the P450 CYP3A4 enzyme is responsible for N-dealkylation [425]. Clearance in neonates undergoing cardiovascular surgery (6.7 SD 6.1 mL/kg/min) is reduced compared with values of 18.1 SD 2.7, 16.9 SD 3.2 and 13.1 SD 3.6 mL/kg/min in infants, children and adolescents, respectively (Table 25.7) [423], consistent with rapid development of hepatic metabolic pathways. Clearance maturation standardised to a 70 kg person using allometry is similar to that of other drugs that depend on CYP3A4 for metabolism (e.g. levobupivacaine, fentanyl, alfentanil) [426]. Similarly, clearance in infants (27.5 SD 9.3 mL kg−1 min−1) was greater, expressed as per kilogram, than those in children (18.1 SD 10.7 mL kg−1 min−1) in another study of children undergoing cardiovascular surgery [427]. Clearance in healthy children (2–8 years) was greater (30.5 SD 8.8 mL kg−1 min−1) than those undergoing cardiac surgery [424]. The elimination of sufentanil is unaffected by renal failure but markedly altered by factors that influence hepatic blood flow (perfusion-limited clearance) [424]; cirrhosis has little effect on its elimination [428].
Table 25.7
Sufentanil PK in children undergoing cardiovascular procedures
Age | Weight (kg) | CL (mL/min/kg) | CL (mL/kg/70 kg) | V (L/kg) | V (L/70 kg) |
|---|---|---|---|---|---|
1–30 days | 3.24 | 6.7 | 218 | 4.15 | 290.5 |
2–23 months | 8.7 | 18.1 | 752 | 3.09 | 216.3 |
3–11 years | 21.0 | 16.9 | 876 | 2.73 | 191.1 |
48–70 years | 58.4 | 13.1 | 876 | 2.75 | 192.5 |
Reported adverse effects are similar to those described for other opioids, bradycardia, nausea and vomiting, chest wall rigidity and respiratory depression.
Intranasal sufentanil may have a role for premedication, procedural sedation and analgesia in children, although data in neonates are lacking [328, 429–431]. The dose of sufentanil that is most effective when administered intranasally is 2–3 mcg/kg [430, 432], although reduced dose can be used if combined with nasal ketamine [328]; this combination also reported fewer adverse effects.
Meperidine (Pethidine)
Meperidine is a weak opioid, primarily μ-receptor, agonist that has a potency of 1/10 that of morphine. The analgesic effects are detectable within 5 min of intravenous administration, and peak effect is reached within 10 min in adults [433, 434]. Meperidine is metabolised by N-demethylation to meperidinic acid and normeperidine. Meperidine clearance in infants and children is approximately 8–10 mL/min/kg [421, 435] Elimination in neonates is greatly reduced, and elimination half-time in neonates, who have received pethidine by placental transfer, may be two to seven times greater than that in adults [436]. The elimination half-life of meperidine in children after IV administration is approximately 3 SD 0.5 h with a variable half-life in neonates between 3.3 and 59.4 h. The V ss in infants, 7.2 (3.3–11) L/kg [421], is greater than that in children 2–8 years of 2.8 SD 0.6 L/kg [435].
The principal current use of meperidine (pethidine, Demerol) in children is to stop postoperative shivering. Although its onset time is more rapid than morphine, there is a risk of seizures after repeated dosing due to accumulation of the metabolite normeperidine (norpethidine) [437]. Clinical use is decreasing as a consequence. The impression that meperidine causes less histamine release than morphine has been questioned [438]. Meperidine is no more effective for treating biliary or renal tract spasm than comparative μ-opioids [439]. The purported benefits of substituting meperidine for morphine in children who are hypovolemic or asthmatic are questionable.
Respiratory depression in infants after meperidine appears to be less than after morphine [91], probably a consequence of an increased V ss in that age group. Meperidine was used for a number of years as a component of various ‘lytic cocktails’ that provided sedation. It was administered either rectally or orally. The safety of these admixtures especially in neonates is dubious, and use is now infrequent [266]. Meperidine’s local anaesthetic properties have been found useful for epidural techniques [440].
Hydromorphone
Hydromorphone is a semisynthetic congener of morphine with a potency of around 5–7.5 times that of morphine [441]. Hydromorphone is metabolised to hydromorphone-3-glucuronide and also, to a lesser extent, to dihydroisomorphine and dihydromorphine [442]. Its bioavailability is about 55 % after nasal and oral administration and about 35 % after rectal administration (not recommended) [443–445]; there is high first-pass metabolism [446]. A clearance of 51.7 (range, 28.6–98.2) mL/min/kg is reported in children [447] with a half-life of 2.5 SD 0.9 h [443–445, 447].
Hydromorphone (Dilaudid) is commonly used when prolonged analgesia is required. Morphine is often changed to hydromorphone to reduce adverse effects or because of concern of accumulation of morphine metabolites, particularly in the presence of renal failure. Hydromorphone is commonly administered IV, orally, in the epidural space and more recently through the nasal mucosa [448, 449]. Hydromorphone is used for chronic cancer pain, and plasma concentrations of around 4.7 ng/mL (range 1.9–8.9 ng/mL) relieve mucositis in children given PCA devices [441, 447].
Oxycodone
Oxycodone is an analgesic with a minimal effective concentration of 25 mcg/L and a target concentration of 45–50 mcg/L for postoperative pain relief [64, 450]. Oxycodone (OxyContin) is another long-acting semisynthetic opioid that is usually administered orally and is available in a controlled-release formulation [451]. The relative bioavailability of intranasal, oral and rectal formulations was approximately 50 % that of intravenous in adults. The buccal and sublingual absorption of oxycodone is similar in young children [452]. The bioavailability after various routes IM is 68 %; buccal, 55 %; and orogastric, 37 % [453]. Oxycodone may also be administered rectally, with a similar bioavailability, although absorption can be slow [454]. The IV formulation of oxycodone significantly depresses respiration. Transmucosal deliver offers another route of administration [455]. Oxycodone (0.1 mg per kg) in children after ophthalmic surgery caused greater ventilatory depression than other opioids [456]. This respiratory depression was also evident in neonates breastfed by mothers treated with oxycodone. Maternal exposure to oxycodone during breastfeeding was associated with a 20.1 % rate of neonate CNS depression compared with 0.5 % in those given acetaminophen and 16.7 % in those given codeine [457].
As with many medications, interindividual variability in the elimination half-life of oxycodone in the neonates and infants is large [458, 459]. In children, the elimination half-life after IV, buccal, IM or orogastric administration is 2–3 h [460]. Oxycodone is metabolised mainly by CYP3A4 with a small contribution from CYP2D6 [461]. Mean values of drug clearance and volume of distribution (V ss) were 15.2 (SD 4.2) mL/min/kg and 2.1 (SD 0.8) L/kg in children after ophthalmic surgery [456].
This opioid is commonly used to transition from patient-controlled analgesia and to treat chronic painful conditions.
Methadone
Methadone is a synthetic opioid with an analgesic potency similar to that of morphine but with a more rapid distribution and a slower elimination. Methadone is used as a maintenance drug in opioid-addicted adults to prevent withdrawal. Methadone might have beneficial effects because it is a long-acting synthetic opioid with a very high bioavailability (80 %) by the enteral route. It also has NMDA receptor antagonistic activity. Agonism of this receptor is associated with opioid tolerance and hyperalgesia. Methadone is a racemate and clinical effect is due to the R-methadone isomer. Methadone is 2.5–20 times more analgesic than morphine [462]. Methadone has high lipid solubility (octanol–water partition coefficient LogP = 3.93), similar to fentanyl (LogP = 4.05) and sufentanil (LogP = 3.95) [109], and such drugs rapidly distribute into fat tissues (including that in CNS).
The primary indication for methadone in children is to wean from long-term opioid infusions to prevent withdrawal [392, 463, 464] and to provide analgesia when other opioids have failed or have been associated with intolerable side effects [465, 466]. Intravenous methadone has been shown to be an effective analgesic for postoperative pain relief [467], and oral administration has been recommended as the first-line opioid for severe and persistent pain in children [468]. It seems also to be a safe enteral alternative for intravenous opioids in palliative paediatric oncological patients [469].
A minimum effective analgesic concentration of methadone in opioid-naïve adults is 0.058 mg/L [470], while no withdrawal symptoms were observed in neonates suffering opioid withdrawal if plasma concentrations of methadone were above 0.06 mg/L [471]. The racemate of methadone which is commonly used in paediatric and anaesthetic care is metabolised to EDDP (2-ethylidine-1,5-dimethyl-3,3-diphenylpyrrolidine) and EMDP (2-ethyl-5-methyl-3,3-diphenylpyrroline).
Methadone is cleared by the cytochrome P450-mixed oxidase (CYP3A4, CYP2B6 and CYP2D6) enzyme systems, all of which are immature at birth [144]. CYP3A7 may contribute to clearance in the neonate [158]. Foetal liver microsomes are reported to show extremely high CYP3A7 levels (311–158 pmol mg−1 protein), and there is significant expression through to 6 months postnatal age; the decrease in activity mirrors the rise of CYP3A4 activity [158]. Methadone has high lipid solubility [472] with a large volume of distribution of 6–7 L/kg in children and adults [473–475]. Pharmacokinetic parameters, standardised to a 70 kg adult using allometry, have been estimated using a three-compartment linear disposition model. Population parameter estimates (between-subject variability) were central volume (V 1) 21.5 (29 %) L 70 kg−1, peripheral volumes of distribution V 2 75.1 (23 %) L 70 kg−1, V 3 484 (8 %) L 70 kg−1, clearance (CL) 9.45 (11 %) L h−1 70 kg−1 and inter-compartment clearances Q2 325 (21 %) L h−1 70 kg−1 and Q3 136 (14 %) L h−1 70 kg−1. EDDP formation clearance was 9.1 (11 %) L h−1 70 kg−1, formation clearance of EMDP from EDDP 7.4 (63 %) L h−1 70 kg−1, elimination clearance of EDDP 40.9 (26 %) L h−1 70 kg−1 and the rate constant for intermediate compartments 2.17 (43 %)/h [143]. These parameter estimates in children and neonates are consistent with those reported by others in neonates [476], children [475], adolescents [477] and adults [478]. There was no clearance maturation with age (Fig. 25.16). Neonatal enantiomer clearances were also similar to those described in adults [143].
A IV regimen of 0.2 mg kg−1 per 8 h in neonates achieves a target concentration of 0.06 mg L−1 within 36 h. Infusion, rather than intermittent dosing, should be considered if this target is to be achieved in older children after cardiac surgery. Analgesic response in adults on chronic methadone programmes suggests a steep concentration–response relationship for pain relief (Hill = 4.4 SD 3.8) with very rapid equilibration between plasma methadone concentrations and the sites mediating pain relief [479]. Consequently, the drug rapidly loses effect as concentrations fall below the EC50. A single dose of 0.2 mg kg−1 will contribute little analgesia after a few hours. An infusion (Fig. 25.33) has been suggested for analgesia in teenagers after spinal instrumentation surgery [477], and consideration could be given to this technique after cardiac surgery in children.
Fig. 25.33
The upper panel shows a simulation for a 3.5 kg neonate given a methadone loading dose of 0.6 mg kg−1 followed by a maintenance dose of 0.15 mg kg−1 8 h. EDDP concentrations track parent drug concentrations. The methadone target concentration of 0.06 mg L−1 is achieved rapidly when compared to the neonate given 0.2 mg kg−1 8 hourly without a loading dose. A single dose of methadone 0.2 mg kg−1 given for postoperative analgesia in a child is unlikely to achieve long duration of analgesia because concentrations are below 0.03 mg kg−1 within 1.5 h (lower panel). Infusion may be a better option. A regimen comprising methadone bolus 0.15 mg kg−1 followed by 0.15 mg kg−1 h−1 for 1 h, 0.075 mg kg−1 h−1 for 2 h and 0.025 mg kg−1 h−1 for 6 h maintains a concentration of 0.06 mg L−1. EDDP concentrations after this infusion regimen are also shown. (Reproduced from Ward RM, Drover DR, Hammer GB, Stemland CJ, Kern S, Tristani-Firouzi M, Lugo RA, Satterfield K, Anderson BJ. The pharmacokinetics of methadone and its metabolites in neonates, Infants and children. Pediatr Anesth 2014;24: 591–601 [143], with kind permission from John Wiley and Sons)
Drug-induced prolongation of the QT interval is a recognised risk factor for ventricular arrhythmias. While methadone is known to prolong the QT interval, the effect of methadone on QTc is associated with chronic dosing in adults rather than an isolated dose, and it is the daily methadone dose that correlates positively with the QTc interval [480, 481].
Codeine
Codeine, or methylmorphine, is a morphine-like opioid with 1/10 the potency of morphine. It is mainly metabolised by glucuronidation, but minor pathways are N-demethylation to norcodeine (10–20 %) and O-demethylation (CYP2D6) to morphine (5–15 %) [170]. Although maturation of codeine clearance with age has not been reported, it is expected to follow that of morphine because glucuronidation is the major pathway, where maturation is mostly complete by 1 year of age [45].
Approximately 10 % of codeine is metabolised to morphine. As the affinity of codeine for opioid receptors is very low, the analgesic effect of codeine is mainly due to its metabolite, morphine [170]. Codeine is effectively a prodrug analgesic. There is little evidence for the broad belief that codeine causes fewer adverse effects, such as sedation and respiratory depression, compared with other opioids [170]. The continued use of this minor opium alkaloid for paediatric analgesia remains baffling and is subject to debate because it is a prodrug [482].
The primary routes for delivery of codeine are oral and IM. Intravenous codeine was used in the past, but serious life-threatening adverse effects including transient but severe cardiorespiratory depression [483–485] and seizures [486] led to proscription of this route of delivery.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


