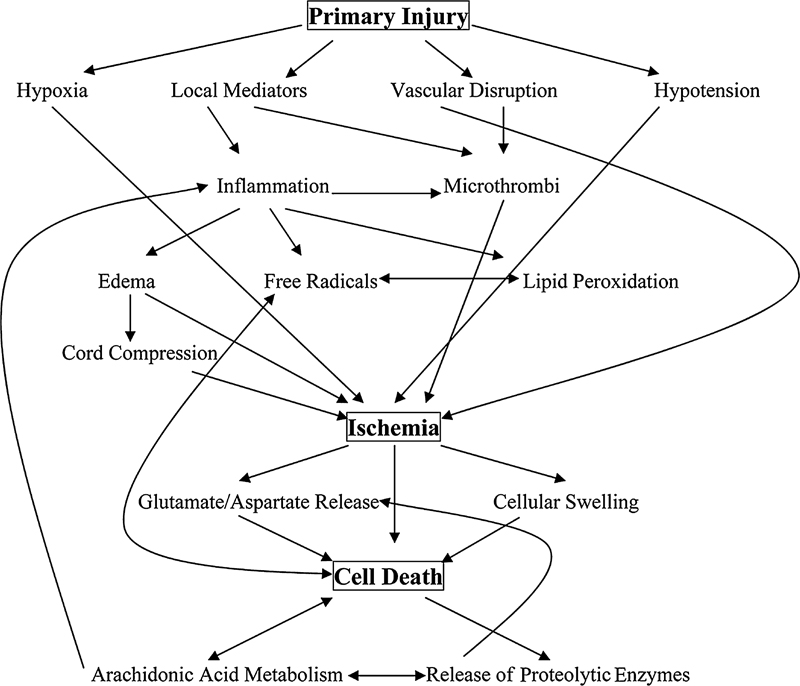
2 Pathophysiology of Spinal Injuries Jeff S. Silber, Adam S. Levin, Michael Kessler, and Kamal D. Dagly There are approximately 12,000 new cases of spinal cord injury (SCI) reported annually in the United States, and there are approximately 200,000 to 250,000 patients with various spinal cord injuries. Most involve males (82%) between 16 and 30 years of age and occur in the summer months on weekends involving motor vehicle accidents. The most common spinal level of injury reported is C5. SCIs produce devastating disabilities for the individual, the family, and society as a whole. SCI injury produces a decreased quality of life as well as a tremendous public health impact, with millions of dollars spent annually caring for these individuals along with lost productivity in this young affected population. The spinal cord is a viscoelastic structure that has the potential to elongate at low applied tensile loads. It is protected by dura, cerebrospinal fluid, and epidural fat, and it is one of only two anatomical structures (the other is the brain) that is entirely encased and protected by bone. The vascular supply is mostly via segmental vessels through radicular arteries. There is an anterior and posterior spinal artery that runs longitudinally along the ventral and dorsal aspect of the spinal cord, respectively, supplying the entire spinal cord. There is a “watershed” area of blood supply located in the midthoracic level that is prone to ischemia in cases of hypotension and decreased blood flow. Fig. 2.1 Flow chart of the primary and secondary spinal cord injury pathways. Spinal cord injury can be divided into two separate injury events known as a primary and secondary injury (Fig. 2.1; Table 2.1). The primary event involves the immediate changes that take place following the initial SCI. Once the initial mechanical injury takes place (motor vehicle accident, diving injury), the subsequent events involve reduction of arterial blood flow to and venous flow from the cord, decreased pO2 supply, decreased Na+-K+ adenosine triphosphatase (ATPase) activity, capillary vessel rupture, electrolyte shifts within the neural tissues, and cell membrane disruption with inflammatory cascades initiated. Following the initial primary injury is a secondary injury cascade. The secondary injury cascade overlaps the primary injury and is more of a continuum of the primary injury. During the secondary injury, there is edema formation within and around the SCI with loss of cellular and blood supply autoregulation. There is an overproduction of opiates, calcium ions, free radical production, excitatory amino acid release, and infiltration of polymorphonuclear leukocytes (PMNs) into the injured area. Table 2.1 Primary and Secondary Spinal Cord Injury Mechanisms
 Primary and Secondary Injury
Primary and Secondary Injury
| Primary injury |
| • Primary injury entails immediate changes following the initial SCI |
| • Once the initial mechanical injury takes place (motor vehicle accident, driving injury), subsequent events involve |
| • Reduction of arterial blood flow to and venous flow from the cord |
| • Decreased pO2 supply |
| • Decreased NA+-K+ ATPase activity |
| • Capillary vessel rupture |
| • Electrolyte shifts within the neural tissues |
| • Cell membrane disruption with initiation of inflammatory cascades |
| Secondary injury |
| • Secondary injury cascade overlaps the primary injury |
| • Events involve |
| • Local disruption of blood flow |
| • Edema formation within and around the SCI |
| • Loss of cellular and blood supply autoregulation |
| • Overproduction of opiates, calcium ions, free radical production |
| • Excitatory amino acid release |
| • Infiltration of polymorphonuclear leukocytes (PMNs) into injured area |
| • Hemorrhage/thrombosis |
| • Axonal degeneration/demyelination |
| • Cell death |
A variety of pathophysiologic processes account for the secondary mechanisms of SCI: edema, inflammation, free radical production, hemorrhage/thrombosis, axonal degeneration/demyelination, and cell death. Nearly all of these phenomena may be either directly or indirectly related to local ischemia at the site of injury. In experimental spinal cord models, ischemia appears to be more detrimental to the gray matter than the surrounding white matter, presumably due to increased metabolic demands of the gray matter. This ischemia can eventually spread vertically in the rostral and caudal directions from the site of initial injury.
The sequence of secondary events begins with a local disruption of blood flow, either as a result of direct microvascular trauma or from microthrombi at the site of initial SCI. As a result, local edema and release of inflammatory compounds from the structures of the damaged cells ensue. Reperfusion injury occurs as a result of the reintroduction of oxidative materials after a period of anaerobic metabolism, leading to a release of free radicals at the site of damage. All of these mechanisms further feed back on themselves, increasing the localized cell death through both necrosis and apoptosis, causing a further leakage from capillaries, an augmented release of cellular breakdown products and inflammatory/degradative enzymes. Significant research has been focused on the cascade of events involved in secondary SCI, as well as means of preventing further destruction. It is our increased understanding of these primary and secondary injury cascades that many therapeutic strategies target in an attempt to alter and decrease the potential negative effects and promote the potential positive effects involved in this complicated injury sequence.
 Phases of Spinal Cord Injury
Phases of Spinal Cord Injury
Spinal cord injury sequences can also be described in a temporal model known as phases. There are three phases described as the SCI site progresses over time: early, intermediate, and late (Table 2.2). During the early phase, from the initial traumatic event, central punctate hemorrhages develop with formation of edema within the spinal cord. Progressive blood vessel disruption occurs with thrombosis, further deceasing blood supply. Axon disruption begins with eventual neuron chromatolysis and cell death. Once this occurs, within days to weeks the intermediate phase continues with progression and longitudinal spreading of the edema in a caudad and cephalad direction within the spinal cord. There is an increased infiltration of PMNs with phagocytosis of the damaged neural tissue. Wallerian degeneration of the axons takes place, and granulation tissue formation starts to develop in place of the native neural tissue. In areas where granulation tissue has yet to develop, cystic cavities form from the phagocytic digestion of the damaged neural cells. The late phase takes place weeks to months following the initial injury. During this phase granulation tissue matures into scar formation, known as gliosis, and large cyst and syrinx formation takes place at and adjacent to the SCI site, progressing longitudinally along the spinal cord. During this phase limited axonal sprouting occurs from the damaged ends of the injury, although there is a progressive loss of white and gray matter with atrophy of the spinal cord. It is the better understanding of these SCI phases that many therapeutic interventions target, and they are discussed below.
Table 2.2 Temporal Model/ Phases of Spinal Cord Injury
| Early phase |
| From the initial traumatic event: |
| • Central punctate hemorrhages develop with formation of edema within the spinal cord |
| • Progressive blood vessel disruption occurs with thrombosis, further decreasing blood supply |
| • Axon disruption begins with eventual neuron chromatolysis and cell death |
| Intermediate phase |
| Within days to weeks after the initial event: |
| • Progression and longitudinal spreading of the edema in a caudad and cephalad direction within the spinal cord |
| • Increased infiltration of PMNs with phagocytosis of damaged neural tissue |
| • Wallerian degeneration of axons |
| • Granulation tissue formation starts to develop in place of native neural tissue |
| Late phase |
| Weeks to months following initial injury: |
| • Granulation tissue matures into scar formation known as gliosis |
| • Large cyst and syrinx formation takes place at and adjacent to the SCI site, progressing longitudinally along the spinal cord |
| • Limited axonal sprouting from the damaged ends of injury |
| • Progressive loss of white and gray matter with atrophy of the spinal cord |
Demyelination/Remyelination
Once the spinal cord undergoes the initial trauma, oligodendrocytes suffer an apoptotic death, and demyelination accompanies wallerian degeneration. It is thought that the primary insult leading to the cascade of events associated with SCI revolves around the local vasculature. Decreased oxygen perfusion, edema, and the influx of inflammatory cells are related to the traumatic breakdown of the blood-spinal cord barrier.1 Key inflammatory cells including the macrophage and activated microglia are found in close proximity to the traumatic lesion and segments of the spinal cord undergoing wallerian degeneration.2 The microglia may lead to oligodendrocyte apoptosis via inducible nitric oxide synthase and reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, both at the injury site and distally.3 Demyelination and the subsequent delay in transmission of neural impulses are the key factors related to SCI patients’ delayed physical advancement and functional recovery.
The traumatic SCI is only the first step in the loss of function. This initial attack on neuron integrity causes local axonal injury and oligodendrocyte death. As the wallerian degeneration occurs, distal axons succumb to the lack of nutrients and progressive demyelination.4 As shown by Crowe et al,5 it is likely that the oligodendrocytes are more susceptible to a traumatic injury, rather than the axons, because oligodendrocyte cell bodies are in closer proximity to the area of trauma, whereas the cell bodies of the axons may be significantly farther away. Once these axons have lost their myelin, they are rendered incapable of conducting action potentials, leaving distal motor units nonfunctioning. In the areas most distal to the site of injury, there is a greater quantitative loss of axons compared with glial cells. This is likely because the oligodendrocytes undergo apoptosis, whereas the axons degenerate and die due to necrosis.6
The loss of axons leads to a decrease in gray matter and the eventual creation of a glial scar at the site of injury, eventually forming a fluid-filled cyst. This cyst has a rim of intact axons that progressively become demyelinated. The cystic cavitation then advances symmetrically both rostrally and caudally from the initial area of injury.6 The advancement of this central cavitation rostrally and caudally causes a significant hindrance to the benefits of therapeutic neural tissue transplantation at the site of injury.
Once the initial injury period has ended, progenitor cells, located at the site of the initial insult, begin remyelinating local axons. Because demyelinated areas lack migratory signals or matrix metalloproteinases, it is unlikely that distant progenitor cells will receive the proper signals to migrate to the injured area. Transplanting myelin-producing cells such as Schwann cells, or olfactory ensheathing glia, has been proposed; however, various complications including graft rejection have yet to be solved.7,8
Axonal Sprouting
Following a traumatic SCI, it is rare for the entire local population of axons to become completely devitalized at the injury site. Some axons remain viable, with the possibility of collateral axonal sprouting with potential filling of the void created during the trauma.9 Unfortunately, there is no guarantee, even if the primary and collateral axons sprout appropriately and are nourished to grow, that the motor and sensory connections will be reestablished. This potential disconnect occurs from the traumatic event, precluding collateral axonal sprouting, and the myelin-associated growth inhibitor Nogo-A has been shown to inhibit growth-promoting proteins in the spinal cord.10,11
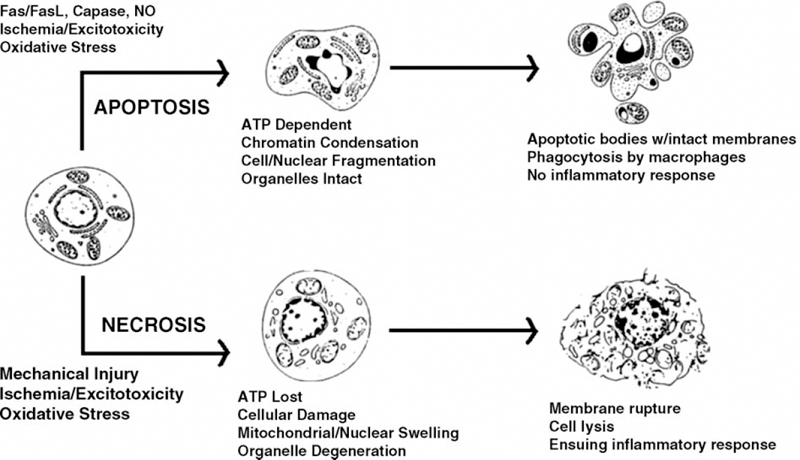
Fig. 2.2 Diagram comparing the similarities and differences between spinal cord cell necrosis and apoptosis.
Although axonal sprouting and the formation of new neural connections distal to the site of injury are the goals after SCI, there are consequences. Aberrant sensory connections have the ability to form that lead to patients suffering from allodynia and various other forms of potentially devastating chronic pain symptoms. Sixty percent of patients with SCI suffer from some type of chronic pain and state that this pain is their primary complaint even if motor function is severely limited.12 Furthermore, autonomic dysreflexia affects over half of the patients with chronic spinal cord injuries that occur at or above the T5-T6 level.13
Future work on the treatment of SCI will focus on reducing inhibitory molecules at the glial scar injury site, leading to increased sprouting from the affected axons. Also, having the ability to focus axonal sprouting toward motor neurons that will make functional connections distally and limit the sprouting of sensory fibers may prevent chronic pain syndromes associated with SCI while maximizing functional recovery.
Apoptosis and Necrosis
Table 2.3 Apoptosis and Necrosis
| Necrosis |
| • Unintentional cell death |
| • During the process of necrosis: |
| • Cells swell passively |
| • Cells lose the potential to create energy |
| • Cells suffer intense mitochondrial damage |
| • Cells lose normal cell hemostasis |
| • Intense secondary inflammatory reaction following cell membrane rupture and cell lysis |
| • Characterized by loss of ATP production |
| Apoptosis |
| • Programmed cell death |
| • Active programmed pathway in neuronal death, akin to suicide, where cell actively kills itself |
| • During the process of apoptosis: |
| • Cells and nuclei characteristically shrink |
| • Cellular organelles stay intact |
| • Nuclear chromatin condenses |
| • DNA fragmentation occurs |
| • No inflammatory reaction because cells are autodigested and phagocytosed in a controlled, organized, and programmed sequence of events |
| • Characterized by ATP-dependent synthesis of proteins, which is required to carry out apoptosis |
Spinal cord cells may undergo death by two mechanisms: (1) unintentional cell death or necrosis and (2) programmed cell death or apoptosis (Fig. 2.2; Table 2.3). Severe cell damage leads to cell death by necrosis, in which the cells passively swell, lose the potential to create energy, suffer intense mitochondrial damage, and lose normal cell homeostasis. Following cell membrane rupture and cell lysis, an intense secondary inflammatory reaction ensues in response to the chemical release from the intracellular structures.14 Apoptosis in contrast to necrosis is an active, programmed pathway in neuronal death, akin to suicide, where the cell actively kills itself. In apoptosis, the cells and nuclei characteristically shrink, cellular organelles stay intact, nuclear chromatin condenses, and DNA fragmentation occurs. Both apoptosis and necrosis can be triggered by exogenous factors such as excitoxins, hyperthermia, free radicals, and radiation. However, cells undergoing death by apoptosis do not produce an inflammatory reaction because they are autodigested by proteases and phagocytosed in a controlled, organized, and programmed sequence of events. Necrosis is characterized by the loss of adenosine triphosphate (ATP) production and energy failure, whereas ATP-dependent synthesis of proteins is required to carry out apoptosis. Although apoptosis and necrosis appear to be distinct and separate pathophysiologic pathways, it is likely that cell death actually occurs along a spectrum between the two mechanisms; however, a distinction is clinically relevant because there is increased potential for therapeutic intervention in the apoptosis pathway.
Emery et al15 confirmed the existence of apoptosis after traumatic human SCI. Data from rat models of SCI have strengthened the notion that apoptosis significantly contributes to SCI and may occur for several weeks after the initial injury at locations that are significantly distant from the site of injury.16–18 All cell types within the spinal cord, including axons/neurons, microglia, oligodendrocytes, and astrocytes, have demonstrated the ability to undergo apoptosis. Apoptosis in microglia leads to secondary injury by means of an inflammatory process.19 Following the first several weeks after acute SCI, oligodendrocytes may begin to express receptors such as Fas and p75, initiating apoptosis and leading to postinjury demyelination. Neuronal apoptosis leads to cell loss that has a profound negative effect on long-term functional outcome and occurs via extrinsic and intrinsic pathways.20 The extrinsic pathway (receptor-dependent) is mediated by Fas ligand and the Fas receptor or by inducible nitric oxide synthase production by macrophages.21,22 This pathway is called the extrinsic pathway because it is activated by extracellular signals, mainly tumor necrosis factor, which is known to rapidly accumulate in the injured spinal cord and activates the Fas receptor. The intrinsic (receptor-independent) or caspase-mediated pathway occurs via direct caspase-3 proenzyme activation or mitochondrial damage, release of cytochrome c, and activation of the inducer caspase-9.23,24 This intrinsic pathway is activated by intracellular signals. Current investigations are focusing on the intrinsic pathway specifically targeted at the caspase inhibitors as a potential therapeutic intervention against this known secondary SCI mechanism.
 Types of Spinal Cord Injury
Types of Spinal Cord Injury
Complete Spinal Cord Injury
Complete SCI is defined as no motor or sensory function below the level of the spinal cord lesion after the resolution of spinal shock (Table 2.4). The usual prognosis of motor or sensory function is poor, and improvement may occur in one or two spinal levels. Spinal shock is defined as a state of physiologic spinal cord dysfunction with no motor or sensory function or autonomic reflexes. This state usually ends within 24 to 72 hours after the initial injury, with return of the most distal innervated reflex known as the bulbocavernosus reflex. The bulbocavernosus reflex is elicited by manually squeezing the glans penis or clitoris or tugging on a urinary catheter with reflex contraction of the anal sphincter. The entire reflex is innervated through the lowest sacral nerve roots, suggesting that spinal shock is over, as the distal end of the spinal cord is functioning enough to allow the reflex to return.
Incomplete Spinal Cord Injury
Table 2.4 Types of Spinal Cord Injury
| Complete |
| • Defined as no motor or sensory function below the level of the spinal cord lesion after the resolution of spinal shock |
| • Usually prognosis is poor, and improvement may occur in one or two spinal levels |
| Incomplete |
| • Defined as any remaining spinal cord tract function after the resolution of the spinal shock |
| • Remaining function may be sacral sparing or any distal sensory of motor function |
| • Several incomplete SCI symptoms include |
| • Central cord syndrome: follows hyperextension injury to the cervical spine with buckling of the ligamentum flavum and posterior vertebral body osteophytes impinging on the cervical spinal cord |
| • Brown-Séquard syndrome: follows injury involving half of the spinal cord, usually from penetrating trauma |
| • Anterior cord syndrome: rare SCI involving anterior two thirds of the spinal cord, leaving only the posterior columns functional |
| • Posterior cord syndrome: isolated injury to the posterior columns |
| • Autonomic dysreflexia: clinical syndrome related to sympathetic nervous dysfunction occurring in up to 85% of patients with a complete SCI above T6 level |
Incomplete SCI is defined as any remaining spinal cord tract function after the resolution of spinal shock (Table 2.4). This remaining function may be sacral sparing or any distal sensory of motor function. Additionally, there are several incomplete SCI syndromes described depending on the anatomical spinal cord area of involvement determining the clinical neurologic presentation. These include the syndromes discussed in the following subsections.
Central Cord Syndrome
Central cord syndrome is seen most commonly in older patients with cervical spondylitic changes resulting in stenosis. It follows a hyperextension injury to the cervical spine with buckling of the ligamentum flavum and posterior vertebral body osteophytes impinging on the cervical spinal cord.
The clinical findings in central cord syndrome consist of motor loss to a greater degree in the upper extremities as compared with the lower extremities with varying degrees of sensory loss below the level of the spinal cord lesion. Urinary retention and sacral sparing may also occur. The somatotopic organization of the spinal cord, with the cervical motor tracts located at a more central location within the spinal cord and the sacral tracts in a more peripheral location, is the anatomical basis for the clinical findings described above (Table 2.5).
Brown-Séquard Syndrome
Brown-Séquard syndrome results from an injury involving half of the spinal cord usually from penetrating trauma. The clinical presentation involves ipsilateral loss of motor function below the level of the lesion (lateral corticospinal tract); ipsilateral loss of fine touch, vibration, and proprioception below the level of the lesion (posterior columns); and contralateral loss of pain and temperature sense one to two levels below the level of the lesion (spinothalamic tract) (Table 2.5).
Anterior Cord Syndrome
Anterior cord syndrome is a very rare SCI involving the anterior two thirds of the spinal cord, leaving only the posterior columns functional. It most commonly occurs following a vascular insult of the spinal cord. Patients usually present with an acute loss of motor and most sensory function below the level of the injury. Sparing of the posterior columns preserves some deep pressure, vibratory, and light touch sensation. The prognosis for recovery is generally poor.
Posterior Cord Syndrome
Isolated injury to the posterior columns is unusual and may present with ipsilateral loss of fine touch, vibration, and proprioception with preservation of pain and temperature sense.
Table 2.5 Incomplete Spinal Cord Injury Syndromes
| Injury/Syndrome | Region Etiology | Clinical Significance |
|---|---|---|
| Anterior spinal cord | Above cauda equina | Ipsilateral: upper motor neuron findings |
| Brown-Séquard | Spinal cord hemisection | Ipsilateral: motor, fine touch, and proprioception loss and contralateral pain and temperature loss |
| Central cord | Cervical spinal cord compression | Upper greater than lower: extremity motor loss, urinary retention, and sacral sparing |
| Autonomic dysreflexia | Bladder/bowel overdistention spinal cord lesion above T6 sympathetic nerves | Hypertension, bradycardia, headache, pupil dilation, and sweating |
Autonomic Dysreflexia
A clinical syndrome related to sympathetic nervous dysfunction is autonomic dysreflexia, also known as hyper-reflexia. This syndrome occurs in up to 85% of patients with a complete SCI above the T6 level. Symptoms include severe headache, sweating, pupillary dilation, bradycardia, and peripheral upper extremity vasodilation. Severe hypertension is the most dangerous manifestation and can lead to convulsions and may even result in intracranial hemorrhage, resulting in death. Autonomic dysreflexia is most commonly due to overdistention of the bladder or bowel viscera that may occur in the SCI patient. Symptoms may begin approximately 4 to 6 weeks after the initial injury. A distended bladder or rectum may cause an abnormal neural discharge of sympathetic afferent nerves, causing reflex activity of the sympathetic efferent outflow. This results in increased sympathetic tone, causing vasoconstriction in the lower extremities, resulting in overall systemic hypertension. The systemic hypertension stimulates the baroreceptors located in the carotid sinus in the neck and aortic arch, causing an increase in central vagal tone as a reaction to try to lower the hypertension. The increased vagal tone results in bradycardia and upper body vasodilation. This response is unable to influence lower body autonomic function because this feedback information cannot descend past the injured spinal cord lesion. This results in a cycle of increased sympathetic discharge and continued extremely elevated systemic blood pressure. Management is rendered on an emergent basis and entails attempts to relieve a distended bladder or impacted bowel. Emergent bowel disimpaction and bladder catheterization along with anti-hypertensive medications are often necessary to control blood pressure and prevent possible recurrence of the condition (Table 2.5).
 Current Therapies (Table 2.6)
Current Therapies (Table 2.6)
Glucocorticosteroids (Methylprednisolone)
One of the most reproducible pathophysiologic changes in SCI is a localized edema and inflammatory response. The exact cascade of inflammatory markers has not been fully elucidated, though many theories have been developed. Since the National Acute Spinal Cord Injury Study (NASCIS)-2 and NASCIS-3 trials, the use of methylprednisolone in the setting of acute SCI has become the standard of care, although recently challenged, with evidence of improvement in long-term clinical outcomes. Proposed mechanisms of action of systemic corticosteroids include a decrease in localized edema, a decrease in microvascular damage with an increase in localized blood flow, stabilization of lysosomal membranes with a resultant decrease in degradative enzyme deposition, a decrease in necrosis in the early and intermediate phases, a decrease in lipid peroxidation and free radical production, and an immune suppression or decrease in inflammatory cytokines25–27 (Table 2.7).
The NASCIS-2 trial was the first trial on human subjects to indicate that pharmacologic intervention after SCI could affect outcomes. The study chose methylprednisolone because of its favorable profile as a corticosteroid that rapidly traverses the blood-brain barrier, with a decreased propensity for causing neutropenia.28,29 It compared SCI patients administered methylprednisolone as a bolus of 30 mg/kg, followed by 5.4 mg/kg/hour over the next 23 hours, to those in a placebo group, as well as to a cohort receiving naloxone. Subjects were further stratified by the timing of initiation of therapy. The study concluded that patients with blunt SCI had improved motor and sensory function at 6 weeks, 6 months, and 1 year after injury in those initiating treatment within 8 hours of injury.28,30
Table 2.6 Summary of Current Therapies
| • Glucocorticosteroids (methylprednisolone) |
| • Other antiinflammatory agents |
| • Lipid peroxidation and reactive oxygen species |
| • Excitotoxicity |
| • Kallikrein |
| • EphA7 |
| • Nitric oxide synthase inhibitor |
| • Inducible nitric oxide synthase (iNOS) |
| • Olfactory ensheathing cells |
| • Stat3/ Socs3 |
| • 4-chloro-3-hydroxyanthranilate |
Table 2.7 Glucocorticosteroids (Methylprednisolone)
| Proposed mechanisms of action of systemic corticosteroids include |
| • Decrease in localized edema |
| • Decrease in microvascular damage |
| • Increase in localized blood flow |
| • Stabilization of lysosomal membranes with resultant decrease in degradative enzyme deposition |
| • Decrease in necrosis in early and intermediate phases |
| • Decrease in lipid peroxidation and free radical production |
| • Immune suppression or decrease in inflammatory cytokines |
After significant criticism of the NASCIS-2 trial, as well as consideration that the proposed effects of glucocorticosteroid therapy would affect components of secondary injury that may endure longer than 24 hours, the role of more prolonged methylprednisolone therapy was studied in NASCIS-3. This trial examined those patients treated with a bolus of 30 mg/kg followed by either 23 or 47 hours of 5.4 mg/kg/hour infusion of methylprednisolone. Findings suggested that for those patients in whom initiation of therapy began between 3 and 8 hours postinjury, the functional outcomes may be improved by prolonging therapy for a total of 48, rather than 24, hours.31 Recently, the usage of methylprednisolone has been intensely challenged and is no longer the standard of care in many SCI centers.
Other Antiinflammatory Agents (Table 2.8)
Glucocorticoid therapy has the potential for a myriad of untoward side effects, including delayed wound healing, systemic immune suppression, gastrointestinal bleeding, and delayed axonal sprouting and repair postinjury. As a result, despite the clinical efficacy of glucocorticosteroids, significant research has focused on potential alternative therapies, with fewer side effects.
Both cyclooxygenases 1 and 2 (COX-1 and COX-2) are capable of converting arachidonic acid into inflammatory prostaglandins. Although both are normally expressed in the spinal cord, and it presently still remains unclear which may play a more important role in inflammation in the diseased state, COX-2 is rapidly upregulated after an SCI.32,33 Studies of animal models have indicated that COX-2 inhibition resulted in improved functional outcomes after injury.34–36
Table 2.8 Other Antiinflammatory Agents
| • Glucocorticoid therapy (side effects include delayed wound healing, systemic immune suppression, gastrointestinal bleeding, delayed axonal sprouting, and repair postinjury) |
| • Both cyclooxygenases (COX-1 and COX-2): recent studies suggest COX-2 inhibition results in improved functional outcomes34–36 |
| • CM101, an antiangiogenic compound |
| • IL-10, an antiinflammatory cytokine |
Additional modalities for prevention of inflammation in the setting of acute SCI have included CM101, an antiangiogenic compound,37 and the antiinflammatory cytokine interleukin-10 (IL-10), which has shown contradictory study results.38,39 Also considered, as in brain injury patients, has been iatrogenic hypothermic therapy either to the entire body or localized to the spinal cord, which has been suggested to decrease edema and metabolic demand on the ischemic spinal cord. This may lead to the decreased release of excitatory amino acids, and ultimately cell death in the acute period after injury. Early trials with small cohorts have indicated favorable results after hypothermia, both systemically and locally.40–43
Lipid Peroxidation and Reactive Oxygen Species (Table 2.9)
Reactive oxygen species as a result of lipid peroxidation has been implicated as a major secondary mechanism of SCI.44 Although the administration of methylprednisolone and other glucocorticosteroids have some action in the inhibition of lipid peroxidation, research into more specific inhibitors, without the adverse reactions of glucocorticosteroids, has shown some promise. Lazaroids, such as tirilizad mesylate, has been shown to have some neuroprotective efficacy with fewer side effects.31,45 The majority of work with these chemicals has been with regard to vascular injury in the brain, but there have been recent encouraging results in spinal injury patients as well.31
Additional compounds such as the immunosuppressive agent cyclosporin A46 and EPC-K1, a phosphate diester linkage of vitamins E and C, have demonstrated efficacy as antioxidants in animal models of SCI.47,48 Melatonin,48 vitamin E, and edaravone, a novel free radical scavenger,49 are additional compounds shown to have equal or superior efficacy compared with methylprednisolone in early animal studies of spinal cord contusions, again without the local and systemic side effects of glucocorticosteroids. Further investigation is required to determine their clinical applicability in humans.
Table 2.9 Lipid Peroxidation and Reactive Oxygen Species
| • Lazaroids, such as tirilizad mesylate: show some neuroprotective efficacy |
| • Immunosuppression agent: cyclosporin A demonstrated efficacy in animal models |
| • EPC-K1, a phosphate diester linkage of vitamins E and C: demonstrated efficacy in animal models |
| • Melatonin, vitamin E, and edaravone, a novel free radical scavenger: demonstrated efficacy in early animal studies |
Excitotoxicity (Table 2.10)
A reproducible pathologic process in the acutely injured spinal cord is the swelling of neurons at the site of injury, presumably from an alteration of ion gradients across the cellular membrane as a result of inflammation and ischemia. These changes in membrane potential, coupled with cellular swelling, lead to a release of excitatory amino acids, namely, glutamate and aspartate. The effects of the release of excitatory amino acids is augmented by an impaired ability for reuptake by the damaged and ischemic cells, resulting in an extracellular increase in glutamate to toxic levels as early as 15 minutes after injury, with levels approaching eight times its normal extracellular concentration compared with normal tissue.50,51
In the brain, it has been shown that intracellular stores of glutamate reach 10 mmol/L, with extracellular concentrations of around 0.6 μmol/L.52–54 Because excitotoxicity can occur with extracellular concentrations of as little as 2 to 5 μmol/L, the potential exists for significant local injury with the death of even a single neuron.50 Presumably, the concentrations in the spinal cord are similar to those measured in the brain with the same potential for excitotoxicity.
| • Changes in membrane potential, coupled with cellular swelling, lead to a release of excitatory amino acids, namely, glutamate and aspartate |
| • This release, combined with an impaired uptake, results in extracellular increase in glutamate to toxic levels |
| • At toxic levels of glutamate, the following occurs: |
| • Massive influx of calcium into neurons |
| • Dysregulation of many cellular processes |
| • Inflammation |
| • Cytotoxicity |
| • Eventually, cell death |
Glutamate acts on the N-methyl-D-aspartate (NMDA), alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA), and kainite receptors on the spinal cord cell surfaces. The NMDA receptor regulates calcium flux across the cell membrane, whereas the AMPA and kainite receptors mediate sodium influx. At toxic levels of glutamate, there is a massive influx of calcium into the neurons, causing a dysregulation of many cellular processes, as well as inflammation and cytotoxicity, resulting in eventual cell death.50 Investigations using antagonists against NMDA receptors, such as MK-80155,56 and gacyclidine,57 and inhibitors against AMPA receptors, such as NBQX,58 have shown improvements in functional outcomes in animal spinal cord contusion models.
Kallikrein (Table 2.11)
Kallikrein 6 (K6) is a member of the kallikrein gene family. The family compromises 15 structurally and functionally related serine proteases. This enzyme is expressed in neurons and oligodendroglia in the adult central nervous system and is also upregulated at sites of SCI. It is hypothesized that K6 may be a key contributor in the pathophysiology of traumatic SCI influencing neural repair and regeneration. Scarisbrick et al59 have shown that in cases of human traumatic SCI, significant increased levels were identified at acute and chronic time frames after the injury. Furthermore, increased K6 levels were identified not only at the injury site, but also at segments above and below. The authors also investigated the potential effects of K6 on neural regeneration by exposing recombinant K6 to laminin, a growth-facilitatory substrate. K6 mediated proteolytic digestion of the growth enzyme, significantly decreasing neurite outgrowth. In contrast, K6 exposure produced hydrolysis of aggrecan, a growth inhibitory substrate, significantly increasing neurite extension. The authors concluded that K6 may influence enzymatic activities involved in secondary SCI, especially axonal outgrowth.
| • Kallikrein 6 (K6): member of the kallikrein gene family |
| • May be a key contributor in the pathophysiology of traumatic SCI, influencing neural repair and regeneration |
| • Significantly increased levels in acute and chronic injury |
| • Increased levels found not only at injury site, but also at segments above and below |
| • Scarisbrick et al59 concluded that K6 may influence enzymatic activities involved in secondary SCI, especially axonal outgrowth |
| • Eph receptor protein tyrosine kinase family and its ligands, the ephrins, have been identified in axonal outgrowth, synapse formation, and target recognition |
| • A recent study demonstrated increased expression of EphA7 1 week post-SCI60 |
| • The study concluded that EphA7 receptors in early stages of SCI pathophysiology (first week) may play a significant role60 |
EphA7 (Table 2.12)
Functional impairment following SCI is partially attributed to neuronal cell death. Further degeneration is seen with apoptosis of central nervous system myelin-forming oligodendrocytes. The Eph receptor protein tyrosine kinase family and its ligands, the ephrins, have been identified in axonal outgrowth, synapse formation, and target recognition. A recent study investigated the effects of EphA7 after SCI.60 The authors demonstrated an increase in the expression of EphA7 1 week postinjury. Receptor immunoreactivity was most prevalent in astrocytes of the white matter in the center of the injury. Furthermore, blocking the expression of EphA7 after SCI resulted in significant acceleration of hindlimb locomotor recovery at 1 week in treated animals, but at 2 weeks there was no difference as compared with controls. The authors concluded that EphA7 receptors in the early stages of SCI pathophysiology (first week) may play a functional role.
Table 2.13 Nitric Oxide Synthase Inhibitor
| • Positive effects of nitric oxide synthase on neuronal survival are well known |
| • Effect on regeneration of neuronal tissue being explored |
| • A recent study concluded that nitric oxide synthase is potentially beneficial to the amount of axonal regeneration in injured spinal cord motoneurons61 |
Nitric Oxide Synthase Inhibitor (Table 2.13)
The positive effects of nitric oxide synthase on neuronal survival are well known. Its effect on regeneration of neuronal tissue is now being explored. A recent study investigated the role of nitric oxide synthase on injured motoneurons in rats.61 The investigators avulsed a ventral motor root from the spinal cord and transplanted a section of peripheral nerve directly into the spinal cord. The results showed that the rats treated with nitric oxide synthase inhibitor reduced the number regenerating motoneurons to 50% at 2 and 4 weeks as compared with sham controls. Only the total number of motoneurons regenerating was reduced as the rate of axonal regeneration of the existing motoneurons was not affected. The authors concluded that nitric oxide synthase is potentially beneficial to the amount of axonal regeneration in injured spinal cord motoneurons.61
Table 2.14 Inducible Nitric Oxide Synthase (iNOS)
| • One of three isoforms of nitric oxide synthase producing nitric oxide |
| • iNOS producing increased amounts of nitric oxide has been found to be a major contributor in initiation and promotion of inflammatory reactions following SCI |
| • On the other hand, complete removal of iNOS and the formation of nitric oxide can be more detrimental than protective |
| • The extracellular environment in which nitric oxide is produced determines whether it is detrimental of advantageous |
Inducible Nitric Oxide Synthase (iNOS) (Table 2.14)
Inducible nitric oxide synthase (iNOS) is one of three isoforms of nitric oxide synthase producing nitric oxide. Inducible nitric oxide synthase producing increased amounts of nitric oxide has been found to be a major contributor in the initiation and promotion of inflammatory reactions following an SCI. Therefore, iNOS and nitric oxide generation has become a therapeutic target in SCI patients. In contrast, it has been shown that the complete removal of iNOS and the formation of nitric oxide can be more detrimental than protective. Additionally, it is now believed that the cellular environment in which nitric oxide is produced determines whether it is detrimental or advantageous. This dilemma makes therapeutic interventions targeted against the formation of nitric oxide with iNOS a bit more complicated. Investigators are now trying to produce a pharmacologic strategy (nitric oxide synthase inhibitor) that, it is hoped, will produce a delicate therapeutic balance and therefore allow the production of a physiologic amount of nitric oxide.62
Olfactory Ensheathing Cells (Table 2.15)
Olfactory ensheathing cells (OECs) have been a recent area of intense interest and study. The cells are located in the olfactory bulb of the nose, and the nerve cells act as a conduit between the peripheral nervous system and the central nervous system. It is this unique anatomical situation that has been the center of interest. The theory is that these cells can be transplanted at an SCI site, aiding axonal growth across the defect. Recently, Moon et al63 reported on the transplantation of OECs following complete spinal cord transaction in rats. The authors reported an induced recovery of hindlimb function 14 weeks postinjury in rats placed in motor enrichment housing, which encouraged hindlimb exercise, as compared with rats placed in a basic housing. which did not.
Table 2.15 Olfactory Ensheathing Cells (OECs)
| • Hypothesis that OEC, located in the olfactory bulb of the nose, can be transplanted at an SCI site, aiding axonal growth across the defect |
| • Lima et al64 concluded that olfactory mucosa autograft transplantation into complete human SCI sites was potentially beneficial |
Lima et al64 reported on seven patients with transplanted olfactory mucosa autografts following a complete SCI. The olfactory mucosal grafts contain olfactory ensheathing and stemlike progenitor cells for neural repair. All patients were American Spinal Injury Association (ASIA) class A and were between 18 and 32 years of age. They underwent an olfactory mucosal autograft transplantation as early as 6 months and as late as 6.5 years after injury. The spinal cord lesions ranged from 1 to 6 cm in length and were present between the C4 and T6 spinal levels. Posttransplantation magnetic resonance imaging (MRI) studies revealed moderate to complete filling of the SCI defects. Two of the seven patients went from ASIA class A to class C, and every patient had improvement in ASIA motor scores. The authors concluded that olfactory mucosa autograft transplantation into complete human SCI sites was potentially beneficial.
| • Over time, reactive astrocytes form a scar that is thought to be detrimental to reformation and regeneration of axonal growth |
| • Astrocytes may play a more crucial positive role following SCI in wound healing and ultimately functional recovery |
| • Stat3: protein signal transducer and activator of transcription 3 |
| • Socs3: protein suppressor of cytokine signaling 3 |
| • Okada et al65 concluded that Stat3 may be a target strategy for a positive intervention following SCI |
Stat3/Socs3 (Table 2.16)
In the injured central nervous system, including the spinal cord, over time reactive astrocytes form a glial scar, which is thought to be detrimental to the reformation and regeneration of axonal growth. Some researchers theorize that astrocytes may play a more crucial positive role following SCI in wound healing and ultimately functional recovery. Okada et al,65 using a mouse model, following an iatrogenic spinal cord contusion selectively deleted either a protein signal transducer and activator of transcription 3 (Stat3) or a protein suppressor of cytokine signaling 3 (Socs3). The mice selectively deleted for Stat3 (without activator) showed limited migration of astrocytes with increased infiltration of inflammatory cells, neural cell disruption, and axonal demyelination leading to severe motor deficits. In contrast, the Socs3 (without suppressor) mice showed rapid migration of reactive astrocytes, decreased inflammatory cells, a smaller contracted contusion area, and improvement of motor recovery and functional outcomes. The authors concluded that Stat3 may be a target strategy for a positive intervention following an SCI.
Table 2.17 4-Chloro-3-Hydroxyanthranilate
| • Secondary mechanisms following SCI involve inflammatory response |
| • One mechanism involves activation of neurotoxins by activated macrophages and microglia |
| • Recently researcher evaluated QUIN (neurotoxin quinolonic acid) in adult guinea pig model |
| • Administration of 4-chloro-3-hydroxyanthranilate (4CL-3HAA) initiated 5 hour after injury attenuated local amount of QUIN production by 50% with reduction in severity of secondary functional deficits |
| • Hartley et al concluded that QUIN contributes to secondary functional deficits and its reduction with 4CL-3HAA can lead to improved functional outcomes following acute SCI |
4-Chloro-3-Hydroxyanthranilate (Table 2.17)
It is well documented that secondary mechanisms following SCI involve an inflammatory response. One mechanism involves activation of neurotoxins by activated macrophages and microglia. Recently, researchers evaluated the role of endogenous tryptophan metabolite and neurotoxin quinolinic acid (QUIN). In an adult Hartley guinea pig spinal cord contusion model, QUIN accumulated at the site of SCI. Furthermore, on postinjury day 12, the amount of QUIN that accumulated was proportional to the severity of the functional neurologic deficits. The administration of 4-chloro-3-hydroxyanthranilate (4Cl-3HAA) initiated 5 hours after iatrogenic injury attenuated the local amount of QUIN production at the site of injury by 50%, with a reduction in the severity of secondary functional deficits. The authors found that cross-sectional microscopic evaluation of the SCI site revealed a 100% increase in surviving white matter tracts (both axons and myelin) as compared with the untreated group. The authors concluded that QUIN contributes to secondary functional deficits and its reduction with 4Cl-3HAA can lead to improved functional outcomes following an acute SCI.66
 Conclusion
Conclusion
Spinal cord injury can lead to devastating loss of function, resulting in paraplegia and quadriplegia. We are continually gaining knowledge in the pathophysiology of the secondary mechanisms of injury and are developing interventions to either promote or diminish various cascades. Presently, there are no ideal or restorative treatments for patients suffering an SCI. Numerous cellular, molecular, and rehabilitative strategies have been used in animal and human studies, with many of them reaching clinical trials. To date there is no one therapy in sight that appears to be a panacea, but a combination of strategies may soon lead to better long-term functional outcomes for different types of SCI.
References
13. Karlsson AK. Autonomic dysreflexia. Spinal Cord 1999;37:383–391
14. Cohen JJ. Apoptosis. Immunol Today 1993;14:126–130
51. Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988;1:623–634
53. Bouvier M, Szatkowski M, Amato A, Attwell D. The glial cell glutamate uptake carrier countertransports pH-changing anions. Nature 1992;360:471–474
< div class='tao-gold-member'>




