ONCOLOGIC EMERGENCIES
ALISA MCQUEEN, MD AND ANDREW E. PLACE, MD, PhD
GOALS OF EMERGENCY THERAPY
Every year in the United States, approximately 12,000 children and adolescents are diagnosed with cancer and 2,300 children die of their disease or from side effects of treatment. This chapter is organized into two major sections. The first section reviews common presenting symptoms and emergency care considerations for a child who presents to the emergency department (ED) with either a new cancer diagnosis or recurrence of disease. This section is organized by diagnosis and by location of malignancy. The second section addresses the management of complications associated with common pediatric malignancies and their treatment.
The general approach to the pediatric oncology patient in the ED is outlined in Table 106.1. When taking a history from a pediatric oncology patient, exploring the perspective of the patient and parents about the potential cause of the problem and how the patient is doing is a crucial step in the evaluative process. A detailed medication history is critical as patients are likely to be receiving multiple medications (e.g., chemotherapy, antibiotics, antiemetics) whose side effects may be contributing to their presenting symptoms. One last introductory principle is to remember the extreme psychosocial stress that a pediatric cancer diagnosis places on a family. The care of these patients requires the highest level of compassion and professionalism.
KEY POINTS
 The differential diagnosis of many common childhood complaints should include malignant processes.
The differential diagnosis of many common childhood complaints should include malignant processes.
 Prompt recognition of oncologic emergencies has been proven to affect outcomes.
Prompt recognition of oncologic emergencies has been proven to affect outcomes.
 Early consultation with a pediatric oncologist is highly recommended.
Early consultation with a pediatric oncologist is highly recommended.
RELATED CHAPTERS
Signs and Symptoms
• Abdominal Distension: Chapter 7
Medical, Surgical, and Trauma Emergencies
• Endocrine Emergencies: Chapter 97
• Hematologic Emergencies: Chapter 101
• Infectious Disease Emergencies: Chapter 102
• Neurologic Emergencies: Chapter 105
• Renal and Electrolyte Emergencies: Chapter 108
SECTION I: INITIAL CARE OF THE CHILD WITH NEW OR RECURRENT CANCER
Childhood cancer can present with nonspecific signs and symptoms that can overlap with those of many childhood illnesses (Table 106.2). Even when the chief complaint is a localized symptom, disseminated disease may be present. Once the diagnosis of cancer is suspected, the child should be referred to a center skilled in the management of childhood cancer. However, supportive care for life-threatening complications may need to be initiated prior to referral. After stabilization, the specific workup, including obtaining tissue for diagnosis, should be carried out under the direction of a pediatric oncologist so that optimal information can be obtained. No patient should be discharged from the ED without a specific plan for definitive diagnosis and management.
The possibility of a cancer diagnosis usually causes fear and distress and requires empathic care and support from the health care team in the ED. The emergency physician should describe the findings and concern about possible cancer to the patient and family. It is appropriate to reassure them that most childhood cancer is curable. Specific details about diagnosis, treatment, and prognosis are best deferred to the pediatric oncologist once definitive information is available.
LEUKEMIA
Goals of Treatment
The primary goal of emergency management of these patients is rapid assessment and correction of hematologic, metabolic, infectious, and cardiorespiratory complications. After a patient has been stabilized, further diagnostic evaluations can be performed.
CLINICAL PEARLS AND PITFALLS
• Automated differentials may count leukemic blasts as either atypical lymphocytes or monocytes so abnormal numbers of these cell types may actually be due to leukemia.
• Avoid aggressive transfusion in severely anemic but stable patients, as this can result in rapid development of pulmonary edema and respiratory failure.
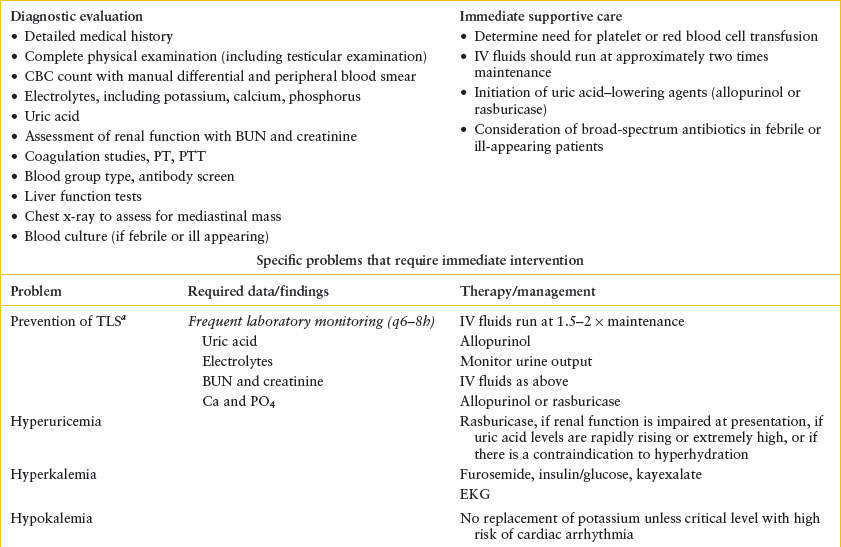
TABLE 106.1
HISTORICAL DATA NEEDED FOR EVALUATION OF PEDIATRIC ONCOLOGY PATIENTS IN THE EMERGENCY DEPARTMENT

Current Evidence
Leukemia is a cancer of white blood cells (WBCs) and their precursors that proliferate in excess within the bone marrow and other hematopoietic tissues. Leukemia is the most common childhood malignancy, accounting for 29% of all cancer diagnoses in children from 0 to 14 years of age. Leukemias are classified as either acute or chronic. More than 95% of pediatric leukemias are acute, with acute lymphoblastic leukemia (ALL) accounting for the vast majority. The remaining leukemias seen in children in order of decreasing frequency include acute myeloid leukemia (AML), chronic myelogenous leukemia (CML), and juvenile myelomonocytic leukemia (JMML). Specific classification is based on morphology, immunologic surface markers, and cytogenetic abnormalities from the bone marrow aspirate. Children with trisomy 21 are at increased risk of transient myeloproliferative disorder of the newborn, AML (younger than 4 years), and ALL (older than 1 year).
Clinical Considerations
Clinical Recognition
The presentations of childhood leukemia are varied and, in some cases, the diagnosis of leukemia is not at all obvious (Table 106.2). Symptoms are usually secondary to bone marrow replacement resulting in cytopenias or from extramedullary infiltration of leukemic blast cells into tissues including the lymph nodes, testes, liver, spleen, central nervous system (CNS), and skin. Systemic symptoms such as fever and weight loss are common.
Patients with acute leukemia are at risk for serious hematologic, metabolic, infectious, and cardiopulmonary complications. The patient with suspected leukemia should be rapidly assessed for evidence of these complications.
Clinical Assessment
Table 106.3 presents assessment and management guidelines for leukemia in the ED. The evaluation should begin with a thorough history and physical examination. The history should focus on the time frame in which symptoms developed and should screen for the complications described above. Assess airway patency, which may be threatened in the setting of a mediastinal mass. Assessment of the patient’s breathing should include attention to the respiratory rate and oxygenation, both of which can become compromised in the setting of anemia, leukostasis, congestive heart failure, and pulmonary infection. In assessing the patient’s circulation, establish intravascular access and include an assessment for evidence of SVC syndrome. The physical examination should include an evaluation for lymphadenopathy and hepatosplenomegaly. Signs of soft tissue infiltration by leukemia cells should be explored, including skin infiltration (leukemia cutis) and testicular enlargement in male patients. A thorough neurologic examination is essential to screen for cord compression and CNS effects of the leukemia. Any abnormalities on neurologic examination warrant further imaging to determine whether a neurologic complication of the leukemia has occurred.
Laboratory evaluation should begin with a complete blood count (CBC), WBC differential, and peripheral blood smear to be reviewed by a hematologist–oncologist or pathologist. Automated differentials might count leukemic blasts as either atypical lymphocytes or monocytes so abnormal numbers of these cell types should raise concern for leukemia. Flow cytometry analysis can provide important diagnostic clues by analyzing the proteins of the blast cell surface. Specific diagnosis requires a bone marrow aspirate, which is not routinely performed in the ED and should be done in consultation with an oncologist.
In constructing a differential diagnosis, it is helpful to consider whether leukemic blasts are present in the peripheral circulation. If blasts are present in substantive quantities (greater than 20%), then leukemia is the most likely diagnosis. A smaller percentage of blasts could indicate a myelodysplastic syndrome, a myeloproliferative disorder, recovery from an aplastic process, or a leukemoid reaction. If blasts are not evident on the CBC, and the patient has pancytopenia, one must consider not only leukemia but also bone marrow failure from aplastic anemia, infection (usually viral), or marrow replacement by a solid tumor. If only one or two cell lines seem to be affected, the clinician should consider the differential diagnoses for each cytopenia individually (see Chapter 101 Hematologic Emergencies).
TABLE 106.2
COMMON PRESENTING SYMPTOMS AND SIGNS OF PEDIATRIC CANCER

TABLE 106.3
EMERGENCY DEPARTMENT CARE OF PATIENT WITH SUSPECTED OR CONFIRMED ACUTE LEUKEMIA

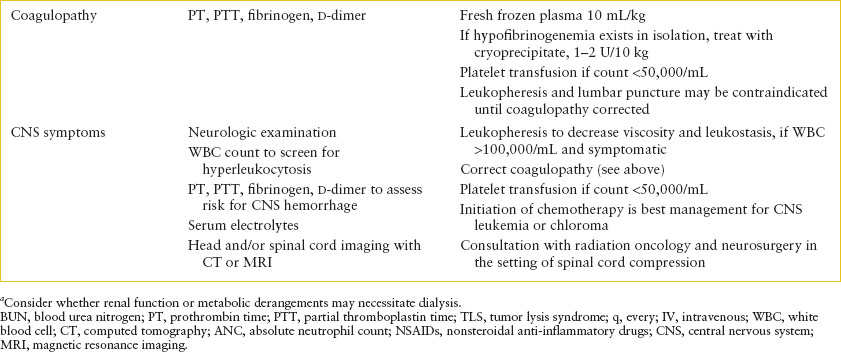
In addition to the laboratory investigations needed for diagnosis, screen for metabolic abnormalities due to tumor lysis by checking serum chemistries, including potassium, calcium, magnesium, phosphorus, and uric acid. Renal function should be assessed with a blood urea nitrogen (BUN) and creatinine. The results of the CBC should be reviewed to assess needs for transfusions of blood products and a prothrombin time (PT) and partial thromboplastin time (PTT) should be checked to look for coagulopathy (Table 106.3). A chest x-ray may indicate the presence of a mediastinal mass or pericardial effusion.
Management
Cytopenias. As the leukemia proliferates in the bone marrow, normal hematopoietic elements decrease, leading to anemia, thrombocytopenia, and neutropenia. This is most common in the setting of leukemia but can occur with solid tumors, such as neuroblastoma and rhabdomyosarcoma with bone marrow metastasis. Anemia can be mild or severe but is often asymptomatic because of its slow development. Anemia with associated clinical signs or symptoms should be treated with a red cell transfusion. Severely anemic but stable patients should be transfused slowly to avoid rapid development of pulmonary edema and respiratory failure. Thrombocytopenia can present with mucocutaneous bleeding such as epistaxis, gingival bleeding, petechiae, and ecchymosis. The risk of bleeding may also be compounded by coagulopathy from the leukemia itself or from disseminated intravascular coagulation (DIC) related to sepsis.
Tumor Lysis Syndrome. Tumor overproliferation or chemotherapy can lead to rapid tumor lysis, in which the release of intracellular contents increases serum levels of lactate dehydrogenase (LDH), potassium, phosphate, and uric acid with potentially severe metabolic consequences. Tumor lysis is common with acute leukemias and lymphomas but can also occur with neuroblastoma or other solid tumors with a very high tumor burden.
Calcium complexes with phosphate to form calcium phosphate crystals that can deposit in the renal tubules and other tissue sites. This can result in renal insufficiency and hypocalcemia. Urate crystals can precipitate in the acidic urine encountered in the renal tubules causing an obstructive uropathy and renal insufficiency. Tumor lysis syndrome (TLS) occurs when these electrolyte derangements occur with evidence of renal insufficiency or failure.
Fortunately, effective preventive strategies make clinically significant TLS a rare occurrence. Screening and preemptive therapy with hydration and allopurinol is appropriate for all patients at risk for TLS. The use of allopurinol or rasburicase (Elitek) to decrease uric acid levels is often driven by institutional protocol. A full discussion of the management of tumor lysis is found in Section II.
Hyperleukocytosis. Hyperleukocytosis is defined as WBC count above 100,000 per mm3. When hyperleukocytosis is present, the clinical findings of leukostasis may develop from sludging of WBCs in the capillary beds. The most vulnerable beds are those in the lungs and CNS where increased viscosity can cause either thrombosis or hemorrhage. Leukostasis is much more common with myeloid leukemia than with ALL. In the setting of hyperleukocytosis, hydration should be initiated immediately to reduce viscosity. Transfusion of red blood cells and diuretics should be avoided to prevent further increases in blood viscosity, but platelet transfusion is appropriate to reduce the risk of CNS hemorrhage. Leukocytopheresis, a technique to reduce blood viscosity acutely, should be initiated immediately in the presence of respiratory or neurologic symptoms, even if mild. The use of prophylactic leukocytopheresis is controversial and should be considered only in consultation with a pediatric oncologist. Since new-onset leukemia may be associated with coagulopathy, we recommend obtaining coagulation studies to determine the risk of bleeding during leukocytopheresis.
Extramedullary Involvement. As leukemia develops, malignant cells may infiltrate nonhematopoietic tissues, producing adenopathy, hepatomegaly, and splenomegaly. Anterior mediastinal masses (AMM) occur primarily with T-lineage ALL and can lead to life-threatening airway compromise and/or superior vena cava (SVC) syndrome. The approach to the presentation and management of AMM is discussed in more detail in the section on thoracic tumors. Chloroma, a mass of leukemic blasts in the soft tissue, can occur in any body part and is much more common with AML than ALL. When this mass develops and rapidly expands near the spinal cord, compression of the cord can result (see “Tumors In and Around the Spinal Cord” section). Leukemic involvement in the spinal fluid can cause meningeal symptoms, cranial nerve palsy, headache, seizure, increased intracranial pressure (ICP), or visual disturbances (from retinal infiltrates). Boys can present with testicular enlargement. There may also be skin lesions due to leukemia cutis (more common in monocytic leukemias and infant leukemia) and gingival hypertrophy (with AML) due to leukemic infiltration.
It is not uncommon for patients with leukemia to be febrile at presentation. Although the fever may be an inflammatory reaction driven by the leukemia itself, serious infection must be explored. It is essential to determine if the febrile patient is neutropenic. If the absolute neutrophil count (ANC) is less than 500 per μL, broad-spectrum antibiotics covering gram-positive and gram-negative bacteria as well as pseudomonas should be administered in the ED. If localizing signs of a bacterial infection are evident, or if high fevers are present (>39°C), empiric therapy should be initiated even if the patient is not neutropenic. Management is summarized in Table 106.3. Patients may present with sepsis at the time of diagnosis or relapse. The increased risk of sepsis may be because of neutropenia and/or immune dysfunction caused by the underlying malignancy. Management of sepsis in the setting of leukemia is not unique and the principles are similar to those described more thoroughly in Chapters 91 Shock and 102 Infectious Disease Emergencies.
Pain. For some patients, limp or a refusal to walk or bear weight is one of the first signs of leukemia. In fact, it is not uncommon for a patient to be treated for diagnoses such as osteomyelitis or septic hip before the diagnosis of leukemia is uncovered. It is essential for the emergency physician to ensure that a refusal to walk is not because of cord compression from chloroma as discussed above. Bone pain is usually due to replacement of the bone marrow with rapidly proliferating leukemic cells causing strain on the marrow spaces. Pathologic fractures may develop as the expanding marrow compartments put strain on and weaken the bony cortex.
HISTIOCYTIC DISEASES
Goal of Treatment
Timely recognition of histiocytic diseases can be lifesaving. The most common histiocytic disease, Langerhans cell histiocytosis (LCH), rarely requires emergency care, whereas hemophagocytic lymphohistiocytosis (HLH) is a life-threatening illness that can be rapidly fatal without appropriate intervention.
CLINICAL PEARL AND PITFALLS
• Consider HLH in the acutely ill infant or young child as these patients can deteriorate quickly and require urgent oncologic consultation and ICU support.
Current Evidence
Histiocytic diseases are a complex and sometimes confusing group of disorders for two reasons. First, several different disease entities make up this group, although efforts have been made to simplify the terminology. Second, significant clinical heterogeneity exists between the major disease entities. Histiocyte is a term referring to several different cells that are thought to derive from a common CD34+ progenitor in the bone marrow. Depending on the cytokines to which the progenitors become exposed, the differentiation can yield tissue macrophages, dermal/interstitial dendritic cells, or Langerhans cells. In general, histiocytic diseases are rare; of the group, the most common is LCH, which has an incidence of three to five cases per million children.
Clinical Considerations
Clinical Recognition
LCH has clinical heterogeneity and the locations involved in the disease have implications for therapy and prognosis. Low-risk LCH may present at any age and systemic symptoms are rare. Skin, bone, lymph nodes, or a combination of these are most commonly involved. Skin involvement can present as a red papular rash, resembling a candidal diaper rash, which may appear on the groin, abdomen, back, or chest. There may also be seborrheic flaking of the scalp, often misdiagnosed as “cradle cap” in infants, draining otitis externa, or ulcerative lesions behind the ears, on the scalp, or in the genital region. Bony involvement may be asymptomatic or painful. A lytic lesion of the skull causing a tender mass is the most common but any bone may be involved. Loose teeth from involvement of the jaw may occur. Certain sites of disease have become known as “risk” organs because their involvement implies more aggressive disease. These patients are often very young with disease that involves the lung, liver, spleen, and/or bone marrow. Lung involvement is rare but worrisome and usually manifests first as hypoxia. Liver and spleen involvement is usually accompanied by enlargement of those organs, although hepatic dysfunction may also be present. Bone marrow involvement is rare, but usually presents with cytopenias, which should prompt a bone marrow aspirate and biopsy. Diabetes insipidus (DI) due to involvement of the posterior pituitary is the most frequent endocrine abnormality in LCH; some patients may present with an apparent “idiopathic” presentation of DI before other lesions are identified. A few patients may present with diarrhea or malabsorption as colitis related to LCH has been described.
HLH is a very rare but severe and life-threatening systemic disease with rapid progression from presentation to death without appropriate intervention. Thus, consideration of this diagnosis in the ED can be critical to outcome. For these reasons, it is essential for the emergency physician to have some familiarity with this disorder. Congenital HLH usually presents in infants and very young children. Other forms of HLH develop secondary to Epstein–Barr virus (EBV) infection, malignancy, or severe rheumatologic disorders or without a specific trigger. Regardless of etiology, HLH presents with fever, hepatosplenomegaly, adenopathy, and rash.
Clinical Assessment
All patients with suspected histiocytic disorders need a thorough history and physical examination as well as a rapid assessment of severity of illness. Patients with HLH may have significant systemic illness with organ dysfunction and even vital sign instability and will often require management in a critical care setting. At the other extreme, patients with suspected localized LCH may require little to no intervention in the ED but need only close oncologic follow-up.
Pulse oximetry should be checked to screen for hypoxia and a chest x-ray obtained if hypoxia is detected. Laboratory evaluation should include CBC, liver function testing, electrolytes to screen for DI, and inflammatory markers such as erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). If HLH is suspected, laboratory analysis may reveal a markedly high serum ferritin as well as transaminitis, hypertriglyceridemia, hypofibrinogenemia, and cytopenias. Bone marrow evaluation may show characteristic hemophagocytosis. Oncologic consultation can guide the evaluation and management of systemically ill patients, as in the case of HLH, or allow for careful follow-up of more stable patients with suspected LCH.
TUMORS OF THE CENTRAL NERVOUS SYSTEM
Goals of Treatment
CNS tumors span a wide range of clinical presentations. The most critical goal is the timely identification and management of cord compression and increased ICP. The goal of early identification of CNS tumors on initial presentation needs to be balanced against exposure to ionizing radiation from CT given that many children present with nonspecific symptoms including headache and/or vomiting. Careful recognition of atypical features and/or concerning associated signs or symptoms can assist in decision making regarding advanced imaging.
CLINICAL PEARL AND PITFALLS
• Clinical clues for increased ICP may include a bulging fontanel in infants, or headache with early morning vomiting in older children.
• Measurement of sodium levels is particularly important in patients with CNS tumors.
Current Evidence
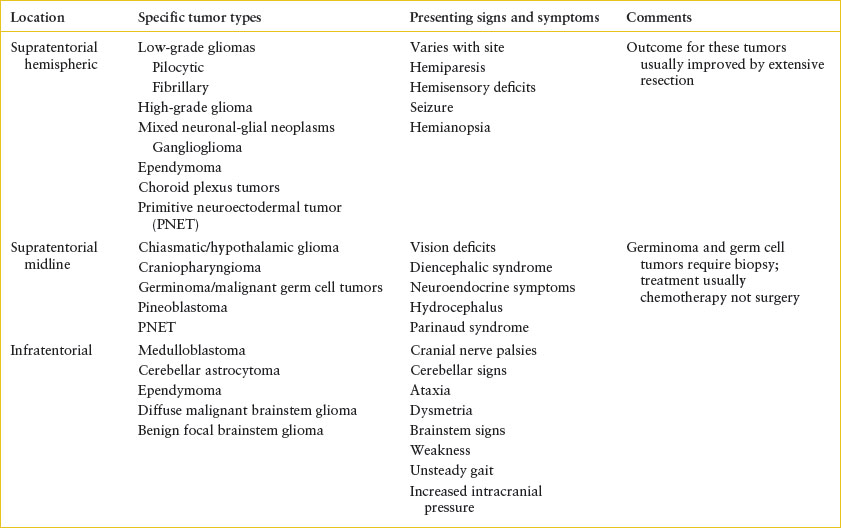
Tumors of the CNS most commonly affect the brain and represent the most common solid tumor in the pediatric population and the second most common pediatric cancer overall. There are approximately 2,000 new malignant brain tumors diagnosed annually in children. These tumors can affect children and adolescents of any age group but the peak incidence is in school-aged children, 5 to 10 years old. Supratentorial tumors are more common in children younger than 1 year and older than 10 years. Infratentorial lesions are more common between ages 1 and 10 years. Unlike in adults, brain tumors in children are usually primary, not metastatic. Since tumor location usually drives the presenting signs and symptoms, this is the most useful categorization in the ED (Table 106.4).
Clinical Considerations
Clinical Recognition
Some of the symptoms of brain tumors in children are nonspecific, and nonlocalizing complaints occur with a variety of tumor types. Examples include headache, altered behavior, vision changes, altered growth or weight, somnolence, and altered school performance. The diagnosis of brain tumor may be delayed in such patients. Once patients develop signs and symptoms more easily referred to the CNS, their presentations tend to hinge on the tumor location (Table 106.4).
Infratentorial tumors may present with cranial nerve deficits, such as facial nerve palsies, dysphagia, or paresis of cranial nerve VI, causing diplopia or strabismus. Ependymomas of the fourth ventricle may present with hydrocephalus and increased ICP. Cerebellar lesions can elicit truncal ataxia and a reeling gait when located on the midline. When only one cerebellar hemisphere is involved, patients may display an ipsilateral hypotonia, resulting in falling to the affected side. Herniation of the cerebellar tonsil can cause head tilt toward the tumor and neck pain or stiffness.
In contrast, supratentorial masses may present with signs and symptoms derived from the involved anatomic locations. Tumors near the optic chiasm may present with vision deficits. Craniopharyngiomas, located in the pituitary gland, can cause visual disturbances, headache, and alterations in the endocrine function. Growth hormone deficiency or hypothyroidism may cause delayed growth. Pineal tumors may cause hydrocephalus by obstructing the aquaduct of Sylvius or can cause Parinaud syndrome (deficits in upward gaze, convergence nystagmus, and impaired pupillary response). Hypothalamic tumors may cause diencephalic syndrome, which includes failure to thrive, wasting, and unusual euphoria. If a tumor is located near the third ventricle, hydrocephalus and symptoms of increased ICP can result. Masses within the cerebral hemispheres themselves usually present with focal motor dysfunction or seizure.
The spinal cord may be the site of a primary tumor or, in some cases, the site of a “dropped metastasis” from a primary lesion within the brain. These can present with focal neurologic deficits attributable to areas of the spinal cord inferior to the lesion (see “Tumors In and Around the Spinal Cord” section).
Brain tumors can cause increased ICP by blocking CSF drainage. The symptoms of increased ICP vary based on patient age. Infants with an open fontanelle can sometimes present with bulging of the fontanelle as well as seizure, vomiting, irritability, or loss of acquired skills. Older children often have headache and early morning vomiting. Sixth nerve palsies are common. Sometimes increased ICP is detected only on imaging of the brain that reveals enlargement of the ventricles or effacement of the gyri.
TABLE 106.4
TUMORS AFFECTING THE CENTRAL NERVOUS SYSTEM
Seizures developing in the setting of a child with cancer, whether due to a CNS tumor or another malignancy, should be managed as described in Chapter 67 Seizures. Brain tumors should be considered in the differential diagnosis of new-onset seizures.
Patients with brain tumors are at risk for several metabolic complications. The presence of an intracranial tumor may be a cause for cerebral salt wasting or SIADH. DI can result from tumor involvement of the pituitary gland. Patients should be screened for these abnormalities with serum chemistries. However, these complications are not managed uniquely because of the brain tumors. Chapter 97 Endocrine Emergencies provides guidelines on evaluation and management.
Clinical Assessment
After ensuring the patient has a stable airway, breathing, and circulation, the evaluation in the ED should focus on a thorough history and physical examination, assessing for any neurologic symptoms and screening for complications described above. A complete physical examination should include a thorough ophthalmologic and neurologic assessment and an evaluation of the patient’s external genitalia for precocious puberty or virilization, since some pediatric brain tumors may be hormone secreting. A rectal examination to evaluate the anal “wink” is also useful as a screen for spinal cord compression. If the history or physical examination raises concern for increased ICP or spinal cord compression, then therapy should be initiated as described below. A CT scan is useful to rule out hemorrhage and assess for increased ICP, and can sometimes be used to visualize a brain tumor. A CT may miss infratentorial masses so in most cases, magnetic resonance imaging (MRI) with gadolinium will ultimately be needed. Laboratory evaluation should include serum electrolytes to evaluate for SIADH, salt wasting, or DI. A CBC is also useful to ensure the patient’s hematocrit and platelet count are adequate for any upcoming procedures.
Management
The emergency management of increased ICP is critical as patients often present with signs and symptoms of this condition at the time of diagnosis. If increased ICP is known or suspected, a lumbar puncture should be avoided, as this theoretically may precipitate a rapid change in ICP followed by herniation of the brain. The pressure may be relieved using steroids such as dexamethasone at a dose of 2 to 4 mg every 6 hours. Mannitol may be useful in decreasing ICP. In some situations, intubation may be needed for both airway protection and to allow for hyperventilation to lower Pco2 when signs of herniation are present. Neurosurgical consultation can address the appropriateness of a ventriculostomy or debulking procedure. The decision about whether to admit a patient with a newly diagnosed brain tumor hinges primarily on the neurologic status. Patients may have altered airway, breathing, or circulation due to the tumor compressing the brainstem. Cranial nerve dysfunction may compromise a patient’s ability to eat normally. The tumor may cause symptoms such as headache, nausea, or vomiting, which require inpatient management. Functional deficits may make discharge problematic. In any of these cases, patients should be admitted to the hospital with prompt consultation with pediatric oncology, pediatric neurology, and pediatric neurosurgery for definitive management.
TUMORS OF THE HEAD AND NECK
Goals of Treatment
Children with head and neck tumors should be rapidly assessed for airway impingement, breathing compromise, and cervical spinal cord compression.
CLINICAL PEARL AND PITFALLS
• Cervical lymphadenopathy is common in childhood and rarely due to cancer. Characteristics that make malignancy more likely include nontender masses, very firm/hard texture, diameter more than 3 cm, adherence to other structures, irregular margins, and absence of signs or symptoms of infection.
• Retinoblastoma often presents with leukocoria (white pupil) first noticed by parents.
Current Evidence
Head and neck tumors represent a diverse range of conditions and, with the exception of retinoblastoma and neuroblastoma, occur most commonly in older children and teenagers. Aerodigestive tract malignancies include sarcomas, lymphoid tumors, and carcinomas. While the latter are commonly seen in the adult population, carcinoma is rare in pediatrics.
Clinical Considerations
Clinical Recognition
Intraorbital tumors may involve any of the tissues contained by the orbit including bone, muscle, soft tissue, and the globe itself. Masses in these regions have a wide differential diagnosis, which includes etiologies such as infections (periorbital and orbital cellulitis), orbital myositis, benign germ cell tumors, or cystic lesions such as a dermoid cyst. Retinoblastoma is the most common intraocular malignancy in children. It occurs in 1 in 23,000 births and is usually diagnosed by age 2. Two-thirds of patients with retinoblastoma present with a white pupil (leukocoria) noted by parents. This is the tumor as seen through the vitreous. The most common malignancies affecting the bony orbit are LCH and neuroblastoma. Presenting symptoms usually include proptosis and strabismus. Vascular tumors including capillary hemangiomas of the orbit may present with red or purple nodular lid lesions or proptosis.
Masses in the aerodigestive tract may be benign, infectious, or reactive in etiology. Regardless of the tissue of origin, these masses usually present with symptoms related to their anatomic position.
Oropharyngeal tumors such as with Burkitt lymphoma can cause snoring and obstructive sleep apnea as well as chronic otitis media and unilateral tonsillar hypertrophy. Gingival hypertrophy may be a sign of a monocytic leukemia. LCH or Burkitt lymphoma of the mandible can present with loose teeth. Rhabdomyosarcoma of the salivary or parotid gland often presents with pain or a facial mass. Malignant tumors of the nose, nasopharynx, and sinuses can present with purulent or bloody rhinorrhea, epistaxis, or sinusitis. Nasopharyngeal carcinoma tends to have a long duration of symptoms before diagnosis because symptoms are rarely specific. Malignant tumors of the sinuses and base of the skull can present with cranial neuropathies such as deviation of the eyes due to compression of the cranial nerves by the tumor. Rhabdomyosarcoma of the middle ear can present with persistent otitis, pain, or cranial neuropathy. The external ear canal can be affected by LCH leading to otorrhea and otitis externa.
Neck masses due to benign congenital anomalies such as branchial cleft cysts or cystic hygromas may grow suddenly as a result of infection or bleeding. Lymphadenopathy in children is common and usually benign. It is most commonly either reactive or infectious in etiology. Bilateral nodes may be associated with viral infections such as EBV or cytomegalovirus (CMV). Unilateral lymphadenopathy, especially in infants and young children, may be associated with Staphylococcus aureus or group A streptococcus infections. Even lymph node enlargement with a chronic time course is still most likely infectious (e.g., mycobacteria, cat-scratch disease, toxoplasma). Lymph nodes can appear large even without infection or malignancy, as observed in Castleman’s, Kikuchi’s, and Rosai–Dorfman syndromes. Enlarged lymph nodes in the neck due to malignancy can be from lymphoma or leukemia or can be because of metastasis from an adjacent solid tumor such as rhabdomyosarcoma, neuroblastoma, or nasopharyngeal carcinoma. It can be difficult to distinguish the primary tumor mass from a lymph node in these circumstances.
Clinical Assessment
Children with neck tumors, regardless of etiology, must be evaluated for impact on the airway, breathing, and circulation. Clinicians should explicitly consider the following:
 Does the mass impinge upon or compress the airway?
Does the mass impinge upon or compress the airway?
 Does the patient experience respiratory distress or is there a compromise to breathing?
Does the patient experience respiratory distress or is there a compromise to breathing?
 Does the mass interfere with circulation of the head and neck leading to SVC syndrome (discussed in section on thoracic tumors)?
Does the mass interfere with circulation of the head and neck leading to SVC syndrome (discussed in section on thoracic tumors)?
 Does the tumor threaten to compress the cervical spine (see “Tumors In and Around the Spinal Cord” section)?
Does the tumor threaten to compress the cervical spine (see “Tumors In and Around the Spinal Cord” section)?
Following this assessment, a careful history should address the duration the mass has been present and its rate of growth, any recent infectious illnesses, the patient’s immunization status, cat exposure, medications, and the presence of systemic systems. Physical examination should screen for other masses or lymphadenopathy in the body. In evaluating nodes of the neck, reactive nodes are often small, mobile, and soft, or while infected nodes may be enlarged, red, and tender. Characteristics that make malignancy more likely are nontender masses, very firm/hard texture, diameter more than 3 cm, adherence to other structures, irregular margins, and absence of signs or symptoms of infection.
Management
Often radiographic imaging and laboratory evaluation is not needed. However, in the case of suspected leukemia or lymphoma, laboratory studies should be obtained to assess for hyperleukocytosis, cytopenias, and TLS (see preceding discussion on leukemia). Radiographic imaging should be pursued if more information is needed about the tumor’s position in relationship to the patient’s airway and other vital structures of the head and neck. A CT scan is often helpful in establishing the neck anatomy in this way. In addition, a chest x-ray should be obtained to explore whether the disease could include a mass in the anterior mediastinum. Laboratory evaluation should include a CBC with differential, ESR and LDH (which may be elevated in certain lymphomas), testing for any relevant infectious etiologies, and consideration of thyroid function testing.
Children with leukocoria should have an urgent ophthalmologic examination to help differentiate retinoblastoma from other possible etiologies such as congenital cataract, coloboma, idiopathic retinal detachment, and others. Direct extension via the optic nerve into the meninges and spinal fluid is a possible but unlikely complication of the tumor. Presentations are usually local and therefore cured by enucleation, but systemic chemotherapy, intra-arterial (ophthalmic artery) chemotherapy, cryotherapy, laser therapy, and insertion of radioactive plaques are all being explored to preserve vision. Management of retinoblastoma hinges on the probability of useful vision in the affected eye. Ophthalmology should be consulted early to determine if the patient’s visual acuity has already been affected by the mass and to plan the urgency of examination under anesthesia. Management of intraorbital tumors may be possible on an outpatient basis, in conjunction with an experienced pediatric ophthalmologist, if the mass is unlikely to affect vision quickly or if vision is already profoundly impaired in the affected eye.
Management of these tumors can sometimes occur on an outpatient basis, in conjunction with a pediatric oncologist and a specialist, such as an oral surgeon or otorhinolaryngologist with expertise in the anatomic region of the tumor. However, specific symptoms such as uncontrolled pain, difficulty maintaining hydration, TLS, or any evolving threat to the airway require inpatient management.
TUMORS OF THE THORAX
Goals of Treatment
The most critical decision making and care in the ED is the differentiation of emergent from nonemergent tumors of the thorax. This difference is frequently driven by tumor location (see Figure 132.11 in Thoracic Emergencies chapter).
CLINICAL PEARL AND PITFALLS
• Children with anterior mediastinal mass must be managed with the utmost caution. Prevention of respiratory failure is critical, as these masses may be located below the carina, rendering even intubation ineffective.
Current Evidence
Thoracic tumors can be caused by a number of childhood cancers. While hematologic malignancies are common, embryonal neoplasms such as neuroblastoma, sarcomas such as primitive neuroectodermal tumor (PNET), and carcinomas can also present in the chest. In general, there are no specific predisposing conditions or factors.
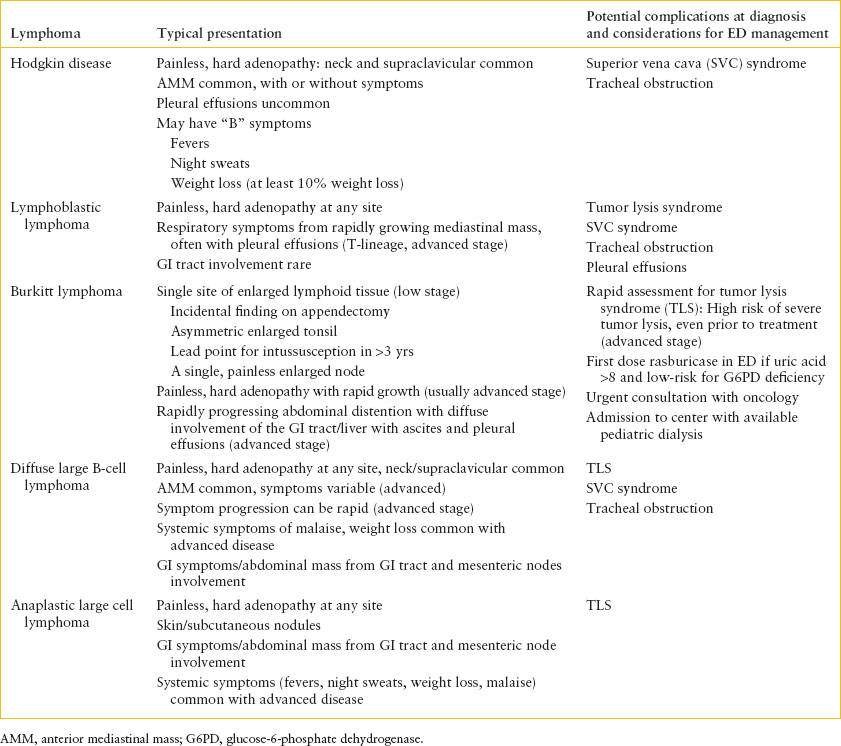
The anterior mediastinum is the most common location of a thoracic tumor in children. The “4 Ts” of the anterior mediastinal tumors include “terrible lymphoma,” “teratoma,” “thymoma,” and “thyroid carcinoma.” The latter three are rare. Nonmalignant conditions with AMM include adenopathy associated with infection, sarcoid, and normal thymus. Common lymphomas (Table 106.5) in the anterior mediastinum include T-cell lymphoblastic lymphoma (or T-cell ALL with an associated AMM), Hodgkin lymphoma, and diffuse B-cell large cell lymphoma. Lymphoma can also occur in the middle mediastinum, which can also be the site of masses associated with pulmonary sequestration and other developmental anomalies. “Teratoma” of the mediastinum includes benign and malignant germ cell tumors. Posterior mediastinal masses include neuroblastoma and other neurogenic tumors such as malignant peripheral nerve sheath tumors (especially in patients with neurofibromatosis, type 1), or benign lesions such as schwannoma.
Primary lung tumors are vanishingly rare in childhood but presentation of lung metastasis at diagnosis or relapse is not uncommon. Many pediatric sarcomas, some lymphomas, germ cell tumors, Wilms tumor, and rarely neuroblastoma can present or recur with lung metastasis. These typically involve multiple small or large lung nodules in the pulmonary parenchyma or are pleural based. Askin tumor is a unique PNET chest wall tumor that occurs in children and young adults.
Clinical Considerations
Clinical Recognition
Tumors in the anterior and middle mediastinum often present with respiratory symptoms ranging from mild cough to severe respiratory distress (Fig. 106.1). These tumors can compress the great vessels and cause SVC syndrome. When asymptomatic they may be identified during evaluation for nonspecific systemic symptoms or even discovered on a chest radiograph performed for another reason. In contrast, posterior mediastinal masses are frequently identified on a chest radiograph performed for another reason. They may, however, cause local pain from nerve root involvement and/or cord compression (see “Tumors In and Around the Spinal Cord” section).

FIGURE 106.1 Chest radiograph demonstrating a large, homogenous anterior mediastinal mass. Patient presented with persistent cough and progressive orthopnea.
TABLE 106.5
LYMPHOMA PRESENTATIONS AND CONSIDERATIONS IN THE ED
The initial complaints associated with pulmonary metastasis may include respiratory insufficiency, postobstructive infection, foreign body–type symptoms, or hemoptysis. Presentation may also involve the discovery of pulmonary nodules on a chest radiograph.
Pulmonary effusions can be the presenting sign of childhood cancer. Effusions can be caused by malignant cells in the pleural space or from obstruction of lymphatic drainage. Effusions are common with AMM due to leukemia or lymphoma and can also occur with posterior mediastinal neuroblastoma, lung metastasis, and chest wall tumors. They may be symptomatic or asymptomatic. The effusion may obscure a mass on both chest x-ray and chest CT scan. Malignant pericardial effusions are most often associated with leukemias and lymphomas. For general diagnosis and management of pulmonary and pericardial effusions see Chapters 94 Cardiac Emergencies and 107 Pulmonary Emergencies. Askin tumor or metastatic sarcomas may present as a chest wall mass with or without pain. More commonly, it presents with respiratory symptoms from an effusion.
Clinical Assessment
There may be useful clues to the diagnosis from the history and physical examination. A recent history of new-onset asthma in an older child who responded to a course of steroids but has now returned is consistent with partial treatment of lymphoblastic leukemia or lymphoma. Many lymphomas can also present with nonspecific systemic symptoms such as weight loss, fatigue, unexplained fevers, night sweats, and malaise. Itching can be a paraneoplastic phenomenon associated with Hodgkin lymphoma. Examination should include assessment of all nodal groups (including axilla and supraclavicular) to both aid in the differential diagnosis and establish a site for possible biopsy. Of note, almost all pediatric lymphomas are high grade and have an acute to subacute course.
The initial focus should include a thorough assessment of airway, breathing, and circulation, all of which may be compromised by an AMM. When an AMM compresses the airway below the level of the carina, intubation will not be effective in managing respiratory failure. Management must focus on prevention of respiratory failure through such strategies as oxygen therapy and maximizing respiratory mechanics. Do not put the patient in a supine position if evidence of respiratory distress. Do not sedate or anesthetize the patient. Do not start empiric steroids without a discussion with an oncologist to ensure that steroids will not interfere with ability to establish the diagnosis. If there is evidence of SVC syndrome (plethora, facial edema, and jugular venous distention), ensure adequate intravascular volume to support systemic return. The patient should not receive anesthesia as it can lead to cardiovascular collapse.
Management
Diagnostic workup in the ED should always include a chest radiograph, including a lateral view, to help establish the location of the mass. Chest CT scan should only be performed in a patient who can comfortably lie supine. CT contrast should not be given without verifying adequate renal function since TLS can occur. Laboratory evaluations should include CBC to assess for evidence of marrow replacement and to identify circulating blasts. Metabolic screening for possible TLS should be performed. Patients with symptoms from an AMM must be admitted to a center with pediatric oncology expertise and may require critical care. If an effusion is drained in the ED for relief of symptoms, a fluid sample should be sent to pathology for cytology if malignancy is suspected. In general, patients who are not hemodynamically compromised by an effusion should not have fluid drained for diagnostic purposes while in the ED.
Patients who are systemically ill require admission for further evaluation and management. Those with minimal or no symptoms from the mass and no metabolic disturbances may be discharged to the care of a pediatric oncologist for further workup as an outpatient.
TUMORS OF THE HEPATOBILIARY TREE
Goals of Treatment
The most critical decision making and care in the ED is the differentiation of emergent from nonemergent. Most patients with newly diagnosed liver tumors in the ED can be managed as outpatients with close subspecialty follow-up.
CLINICAL PEARL AND PITFALLS
• Liver tumors are unusual in childhood and rarely require emergent management, except in the case of severe liver failure or tumor rupture causing hypovolemic shock.
TABLE 106.6
BENIGN AND MALIGNANT ABDOMINAL AND PELVIC MASSES IN CHILDREN

Current Evidence
Tumors of the hepatobiliary tree in children are more likely to be metastases rather than primary tumors of the liver (Table 106.6). Of the primary tumors, hepatoblastoma (HB) and hepatocellular carcinoma (HCC) are the most common (Fig. 106.2). HB usually occurs in patients younger than 6 years and is associated with risk factors such as overgrowth syndromes (e.g., Beckwith–Wiedemann), prematurity, and familial adenomatous polyposis (FAP). In contrast, HCC is more common in patients older than 6 years, particularly in children older than 10 years. Risk factors include chronic liver injury from inborn errors of metabolism such as tyrosinemia, glycogen storage disease type I, chronic hepatitis, chronic iatrogenic androgen exposure, or cirrhosis for any reason. In general, these latter risk factors rarely lead to cancer in childhood.
Clinical Considerations
Clinical Recognition
Typical presenting symptoms include abdominal mass, abdominal pain, and very rarely acute abdomen from massive hepatomegaly or tumor rupture. Other primary liver masses include embryonal sarcoma (extremely rare), nonmalignant vascular lesions such as infantile hemangioendothelioma, and malformations such as hamartoma. Focal nodular hyperplasia can cause a mass on liver imaging but rarely has associated symptoms or hepatic enlargement. Metastatic disease usually is characterized by diffuse nontender enlargement of the liver or multiple small nodules rather than a single dominant mass. Neuroblastoma and advanced hematologic malignancies such as ALL, AML, lymphoblastic lymphoma, and Burkitt’s commonly metastasize to the liver. Although tumors may block biliary drainage, hepatic synthetic function is rarely affected by malignancy in the liver.
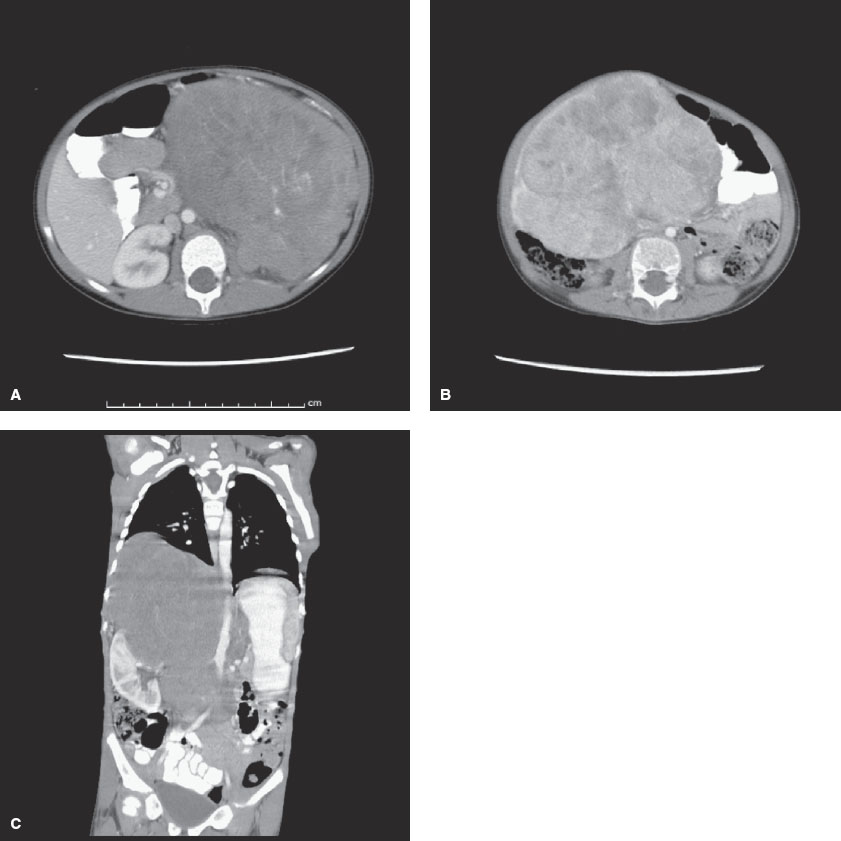
FIGURE 106.2 A: Wilms tumor. Computed tomography (CT) scan of the abdomen reveals a large mass entirely replacing the left kidney. B: Hepatoblastoma. CT scan of the abdomen reveals a large mass arising from the inferior aspect of the liver. The mass enhances heterogeneously with multiple low-density foci. C: Neuroblastoma. CT scan of the abdomen reveals a large mass originating from above the right kidney. The mass crosses the midline and displaces the right kidney laterally and inferiorly. The right adrenal gland is not visualized.
Clinical Assessment
Emergent management is rarely required except in the extremely rare setting of liver failure (see Chapter 99 Gastrointestinal Emergencies), tumor rupture that may require rapid repletion of intravascular volume and blood loss, or severe coagulopathy (see Chapter 101 Hematologic Emergencies). History may elicit risk factors or systemic symptoms such as malaise and anorexia that are more common with HCC than HB. Pain does not help with the differential diagnosis but does require management. Jaundice is most common with HCC but can occur with all liver tumors.
Management
Initial workup should include measurement of AST, ALT, total and direct bilirubin, CBC, PT, PTT, and fibrinogen. Alpha fetoprotein (AFP) can be elevated in both HB and HCC. It is important to note that normal AFP values are high in the first months of life, especially in premature infants. Initial diagnostic imaging should include an ultrasound, which can help identify if a palpable mass is likely to be hepatic in origin and if the liver contains one or multiple masses. If a CT scan is done in the ED, it is important to give intravenous contrast to look for intravascular extension of tumor from the hepatic veins, into the inferior vena cava, and possibly into the right atrium. Renal function should be checked before giving intravenous contrast. Since HB can metastasize to the lungs, consider performing a chest CT scan at the same time in patients younger than 10 years who have a primary liver tumor.
Children who are clinically stable and have a new liver tumor may be discharged from the ED to the care of a pediatric surgeon experienced with liver tumors or a pediatric oncologist. If the patient is unstable, they should be admitted to a center with experience in treating childhood malignancies.
TUMORS OF THE PANCREAS
Pancreatic tumors in children are very rare. They may develop in the setting of a predisposition, such as multiple endocrine neoplasias type 1 (MEN-1) syndrome, which is associated with insulinomas of the pancreas. Insulinomas will present with signs and symptoms of hypoglycemia and a history of “irrational behavior.” Other tumors of the pancreas cause either an abdominal mass or vague, nonspecific abdominal symptoms. The differential diagnosis of a pancreatic mass includes nonmalignant adenoma or cystadenoma as well as malignant entities. Malignant tumors of the pancreas in children may be cystadenocarcinoma, pancreatoblastoma, an embryonal tumor, or an endocrine tumor such as insulinoma, gastrinoma, or VIPoma (Table 106.6). The pancreas may also be affected by metastatic disease from end-stage refractory cancer such as neuroblastoma or rhabdomyosarcoma.
A thorough history and physical examination should assess for endocrinologic ramifications that require medical management, such as hypoglycemia. The evaluation should include a serum AFP, which can be elevated in pancreatoblastoma. Diagnostic imaging may include a CT scan or MRI of the abdomen, but these tests are rarely needed in the ED.
The patient with a newly diagnosed pancreatic tumor may be discharged to home if the patient is otherwise well appearing and if arrangements have been made for an appropriate evaluation, including consultation with a pediatric surgeon, to continue in the outpatient setting. If the patient is ill, or if appropriate follow-up is otherwise unclear, then it is safest to admit the patient to the hospital. Surgical intervention is an important facet of the management plan for patients with pancreatic tumors, as several pancreatic tumors may be managed with just surgery alone.
TUMORS OF THE GASTROINTESTINAL TRACT
Tumors in the gastrointestinal (GI) tract in children include lymphomas, leukemias, gastrointestinal stromal tumors (GISTs), LCH, desmoplastic small round cell tumor, and colorectal carcinomas (Table 106.6). Risk factors for GI lymphomas include primary immunodeficiency. Neurofibromatosis type 1 increases the risk of GIST. FAP, Li–Fraumeni syndrome, and ulcerative colitis increase the risk of colon cancer.
Common presentations include nonspecific symptoms such as weight loss, nausea/vomiting, loss of appetite, change in bowel habits, abdominal distention, or abdominal pain. Chronic GI blood loss can cause iron deficiency anemia. Abdominal distention from masses or ascites may be present. Severe GI bleeding is a rare presentation of GI malignancy but one that requires immediate management as in Chapter 28 Gastrointestinal Bleeding
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


