ONCOLOGIC EMERGENCIES
Children and adolescents develop different types of cancers than adults. The 5-year survival rate for all childhood (0 to 19 years of age) cancers is now >80%1; however, malignant neoplasms remain the second leading cause of death for U.S. children age 5 to 14 years.2 Globally, the American Cancer Society estimates that less than 40% of children younger than 15 years with cancer are adequately diagnosed and treated.3 The most common childhood malignancies are discussed below.
Acute leukemias, including acute lymphoblastic leukemia (ALL) and acute myelogenous leukemia (AML), are the most common cancers in children, accounting for over a quarter of all malignancies.4,5,6 Chronic leukemias are rare in children and adolescents. ALL accounts for approximately 75% to 80% of pediatric leukemias and, if diagnosed early, carries a 5-year survival rate of 90% in developed countries.4,5,6,7 The peak incidence of ALL is 3 to 5 years of age, with younger children having the best outcomes.8,9 In the United States, ALL is more common in boys, and more common in white and Hispanic than in African American children.2,3,4,5,10 A number of inherited risk factors, including trisomy 21, are well documented.
AML accounts for approximately 15% to 20% of childhood and adolescent leukemias in the United States.4,6,11 Cure rates have improved but remain lower than those for ALL. Current estimates are 60% to 70%, and relapsed AML accounts for greater than half of all childhood leukemia-related deaths.4,11,12 Incidence of AML peaks in the first 2 years of life.4,6 Environmental exposures, including chemotherapy agents and radiation received during treatment of other childhood cancers, are known causes of secondary AML. Patients being treated for AML have a higher incidence of complications than those with ALL, especially infections. This is primarily due to the greater intensity of chemotherapeutic regimens necessary to achieve remission.
Most signs and symptoms of acute leukemia are due to bone marrow infiltration by blasts, as well as infiltration of extramedullary sites. A detailed history may reveal nonspecific constitutional symptoms: fever, fatigue, anorexia, and weight loss. Cytopenias from marrow infiltration can present as pallor, easy bleeding/bruising with petechiae and ecchymoses, infections, or bone pain. The reticuloendothelial system is the most common site of extramedullary infiltration, manifesting as hepatomegaly, splenomegaly, and/or lymphadenopathy. Other sites include the CNS and testes.
Consider leukemia in cases of recurrent or severe unexplained epistaxis or other mucosal bleeding or unusually extensive or abnormally located bruising. In rare cases of AML, solid nodules of leukemic blasts (chloromas) may be noted, most commonly in the skin (leukemia cutis) or gingiva. Bone and joint pain may result in limping or refusal to walk. Such symptoms can result in a delay in diagnosis if pain is attributed to other conditions. Concerning symptoms include abnormal blood counts, a history of nighttime pain, and recurrent or persistent bony complaints.13,14 For a subset of patients, diagnosis is made incidentally during an evaluation for infection, lymphadenopathy, priapism, or other presentations.
Begin with a CBC with differential count, a peripheral smear, a coagulation profile, a type and screen, and serum chemistries including calcium, phosphate, magnesium, uric acid, liver function studies, and lactate dehydrogenase. Obtain a chest x-ray, because some children with T-cell ALL can have an anterior mediastinal mass of leukemic cells. If fever or clinical suspicion of infection is present, obtain a blood and urine culture. Although almost all children with leukemia have some form of hematologic abnormality including anemia, thrombocytopenia, leukocytosis, or neutropenia, very few have an extremely elevated WBC. Total WBC counts are often normal or only moderately elevated. Hyperleukocytosis is more likely in patients with AML and in infants with either ALL or AML.15 Definitive diagnosis requires a bone marrow aspirate.
The differential diagnosis of children with marrow suppression includes aplastic anemia, profound iron deficiency anemia (commonly noted in toddlers with excessive milk intake), viral infections (Epstein-Barr virus [EBV], cytomegalovirus [CMV], parvovirus B19), primary immune thrombocytopenia, and rheumatologic diseases. A good rule of thumb is that leukemias commonly involve abnormalities in more than one cell line, whereas other conditions are often restricted to a single cell line.
The goal is to induce remission while minimizing treatment toxicity. Taking into consideration age and leukocyte count at the time of diagnosis, as well as other risk factors (e.g., genetic abnormalities of leukemic cells with important prognostic significance), children with ALL are usually stratified into different treatment groups.16 Children with AML receive intensive multiagent chemotherapy. Molecularly targeted therapeutic approaches are in development.12,17 Some receive hematopoietic stem cell transplantation. Table 143-1 lists common chemotherapeutic agents used in pediatric oncology and their most common adverse effects.
| Chemotherapeutic Agent | Common Indications | Common Adverse Effects |
|---|---|---|
| Doxorubicin (Adriamycin) | NHL, HL, ALL, AML, neuroblastoma, sarcomas, Wilms’ tumor | Myelosuppression, mucositis, cardiomyopathy (acute and chronic), arrhythmia, radiation dermatitis, red urine (from drug), vomiting |
| Bleomycin | Testicular cancer, germ cell tumor, HL | Pneumonitis, pulmonary fibrosis, vomiting, mucositis |
| Carboplatin | Germ cell tumors, sarcomas, neuroblastoma, CNS tumors | Myelosuppression (especially platelets), nephrotoxicity, vomiting |
| Carmustine (bis-chloronitrosourea) | NHL, HL, CNS tumors | Pulmonary fibrosis, stomatitis, myelosuppression |
| Cisplatin | Testicular cancer, germ cell tumor, lymphoma, osteosarcoma, CNS tumors | Nephrotoxicity, myelosuppression, peripheral neuropathy, vomiting |
| Cyclophosphamide | NHL, HL, ALL, neuroblastoma, retinoblastoma, soft tissue sarcoma, Ewing’s sarcoma | Hemorrhagic cystitis, myelosuppression, vomiting, nephrotoxicity |
| Cytarabine (cytosine arabinoside) | ALL, AML, NHL | Myelosuppression, seizure, ataxia/encephalopathy, mucositis, typhlitis, vomiting |
| Actinomycin-D (dactinomycin) | Testicular cancer, sarcomas, Wilms’ tumor | Myelosuppression, veno-occlusive disease, vomiting, mucositis |
| Daunorubicin | ALL, AML | Red urine, myelosuppression, cardiomyopathy, vomiting, arrhythmias, mucositis |
| Etoposide | Testicular cancer, ALL, AML, NHL, HL, CNS tumors, neuroblastoma, sarcomas | Myelosuppression, vomiting, mucositis |
| Ifosfamide | Ewing’s sarcoma, various refractory tumors | Hemorrhagic cystitis, myelosuppression, encephalopathy, nephrotoxicity, vomiting |
| l-Asparaginase; pegaspargase | ALL, AML, lymphoma | Venous thrombosis/cerebrovascular accident, pancreatitis, coagulopathy, platelet dysfunction |
| 6-Mercaptopurine | ALL, NHL | Hepatic dysfunction, myelosuppression, mucositis |
| Methotrexate | ALL, osteosarcoma, NHL | Myelosuppression, renal toxicity, stroke-like syndrome, hepatitis, photosensitivity, mucositis, vomiting |
| Thioguanine | ALL, AML | Myelosuppression, mucositis, veno-occlusive disease |
| Vinblastine | Testicular cancer, HL, histiocytosis | Mucositis, myelosuppression, constipation |
| Vincristine | AML, ALL, HL, NHL, Wilms’ tumor, sarcomas, neuroblastoma | Constipation, peripheral neuropathy, syndrome of inappropriate secretion of antidiuretic hormone/hyponatremia, veno-occlusive disease |
Most children suspected of having leukemia are stable and receive an initial diagnostic workup (see “Diagnosis” section) but no acute treatment. Occasionally, acute treatment is needed for cytopenias, hyperleukocytosis, coagulopathy, electrolyte abnormalities, and infection. Select treatment after consultation with a pediatric oncologist.
Anemia Anemia is extremely common and multifactorial, including impaired hematopoiesis due to leukemic marrow infiltration, decreased erythropoietin levels, iron deficiency, hemolysis, occult blood loss, and chronic inflammation. There is no universally recommended threshold for transfusion, and in the ED, the decision to transfuse should depend more on the appearance and clinical condition of the child than on any particular laboratory value.18 Life-threatening hemorrhage, symptomatic anemia, or rapidly consumptive coagulopathy requires transfusion, with the addition of factor replacement as needed. The general goals of transfusion therapy are to raise the hemoglobin level to >8 to 10 grams/dL, depending on the child’s condition.8 However, in the setting of profound anemia and hyperleukocytosis, transfuse to a much lower level to minimize the effect on blood viscosity. The exact numbers will often depend on institutional protocols (see discussion on transfusion below under “Blood Products”).
Thrombocytopenia Mild to moderate bruising, petechiae, and/or mucosal bleeding may occur with platelet counts <20,000/mm3, but the risk of spontaneous intracranial hemorrhage is extremely low until the platelet count dips to <5000/mm3. Most protocols reserve prophylactic platelet transfusions in asymptomatic patients to platelet counts <10,000/mm3 in the absence of other bleeding risk factors. Invasive procedures require a platelet count >50,000/mm3,8,19 and the platelet count should exceed 50,000 to 100,000/mm3 for surgeries or procedures with bleeding risks.20
Disseminated Intravascular Coagulation Disseminated intravascular coagulation (DIC) is commonly associated with AML. Some subtypes release a procoagulant tissue factor that can lead to a life-threatening consumptive coagulopathy. ED treatment includes replacement of platelets, depleted coagulation factors, fibrinogen with fresh frozen plasma, and cryoprecipitate.
Blood Products A unit of packed red blood cells has a volume of about 250 mL and a hematocrit of 70% to 80%. The typical volume of packed red blood cells given to a child is 10 to 15 mL/kg. Generally, 10 mL/kg of packed red blood cells is expected to raise the hemoglobin level 3 grams/dL in a patient without active hemorrhage.
A dose of platelets is 0.1 unit/kg random donor equivalent units and is expected to raise the platelet count by 30,000 to 50,000/mm3.8,20,21 Some institutions release platelets as single apheresis units versus random donor units. Platelets express ABO antigens, and, even though platelets do not express Rh antigens, Rh-negative donors are used for Rh-negative patients because red blood cell contamination may result in Rh alloimmunization. Rh immunoglobulin can be given if Rh-negative platelets are unavailable. Institutions vary on the use of type-specific platelets and the need for Rh immunoglobulin (Table 143-2).
| Blood Product Component | Volume | Dose | Typical Transfusion Goals* |
|---|---|---|---|
| Packed red blood cells | Citrate phosphate dextrose adenine, 1 unit = 250 mL† | 10 mL/kg increases Hb by 3 grams/dL | Hb, 8–10 grams/dL |
| Adsol, 1 unit = 350 mL† | 12.5–15.0 mL/kg increases Hb by 3 grams/dL | ||
| Platelets | One apheresis unit = 200–400 mL‡ | 0.1 unit/kg increases platelets by 30,000–50,000/mm3 | 50,000–100,000/mm3 |
| Fresh frozen plasma | 1 unit = 200–250 mL (centrifuged) | 20 mL/kg will replace ~50% of most coagulation factors | |
| 1 unit = 500 mL (apheresis) | |||
| Cryoprecipitate | 1 unit = 15 mL | 0.1 unit/kg increases fibrinogen by ~50 milligrams/dL# | Fibrinogen, 100 milligrams/dL |
| 1 unit = 80–100 units factor VIII | |||
| 1 unit = 150–200 milligrams fibrinogen |
There are several product choices for packed red blood cells and platelets: irradiated, leukocyte reduced, and CMV seronegative (Table 143-3). Irradiation of blood products is recommended for profoundly immunosuppressed individuals (e.g., severe immunodeficiency, high-dose chemotherapy, post–bone marrow transplant). Many pediatric cancer centers will transfuse only irradiated blood to all oncology patients. Consult with the pediatric oncologist for specific directions before administering blood products.
| Treatment | Description | Goal | Target Recipient |
|---|---|---|---|
| Leukodepleted blood | 99.9% reduction in donor WBCs | Reduces incidence of febrile nonhemolytic transfusion reactions Decreases sensitization to HLA-1 antigens Significantly reduces transmission of CMV* | Patients likely to receive multiple platelet or packed red blood cell transfusions in future |
| CMV-seronegative blood | Gold standard for CMV-negative blood | Eliminates transmission of CMV* | Patients at high risk for CMV-related complications now or in the future |
| Irradiated blood | Destroys donor lymphocytes’ ability to multiply | Prevent transfusion-associated graft-versus-host disease from donor WBCs (does not prevent CMV transmission) | Profoundly immunocompromised patients Stem cell transplant patients |
Hyperleukocytosis Hyperleukocytosis is a WBC >100,000/mm3. Leukocytosis can cause leukostasis, tumor lysis syndrome, and DIC.22 Leukostasis is a clinical diagnosis. Intravascular aggregations of leukocytes lead to injury of the lungs and CNS most commonly, but can affect other organs as well. In the cerebral circulation, symptoms and signs include headache, mental status changes, visual changes, seizures, and stroke (ischemic and hemorrhagic), whereas in the pulmonary circulation, leukostasis can cause dyspnea, hypoxemia, and respiratory failure. Chest radiographs may be normal or may show a diffuse, nonspecific interstitial infiltrate. AML patients are especially at risk because their leukemic blasts are larger, “stickier,” and more rigid.
ED treatment consists of aggressive IV hydration and treatment of tumor lysis syndrome (see discussion in “Tumor Lysis Syndrome” section and Table 143-5). Avoid treatments that increase blood viscosity such as diuretics and packed red blood cell transfusions. Platelets do not increase blood viscosity and should be administered for levels <20,000/mm3 to decrease the risk of cerebral hemorrhage. Leukapheresis is a temporizing measure until definitive antileukemic therapy can be given.
Hodgkin’s lymphoma is a lymphoid neoplasm characterized by progressive enlargement of lymph nodes. Age distribution is bimodal, with peaks in young adulthood and older-aged adults. It is the most common malignancy in adolescents between the ages of 15 and 19 years old.4 Risk factors include EBV (particularly in developing countries) and human immunodeficiency virus.
Hodgkin’s lymphoma typically presents with painless, firm, “rubbery” lymph nodes, usually cervical or supraclavicular (Figure 143-1). If empiric antibiotics are prescribed for presumptive cervical adenitis, the mass continues to increase in size. The lack of overlying erythema and the absence of pain suggest lymphoma. Systemic “B” symptoms (fever >38°C [100.4°F], night sweats, and weight loss ≥10% over 6 months) are present in 39% to 50% of children and adolescents.23,24 Pulmonary symptoms such as cough, stridor, dysphagia, or dyspnea suggest mediastinal involvement, present in two thirds of pediatric patients at the time of diagnosis.25 Obtain a chest x-ray. Hepatomegaly or splenomegaly can be found due to hematogenous spread. A CT of the soft tissues of the neck, chest, abdomen, and pelvis will help delineate the extent of disease. Subdiaphragmatic primary disease is uncommon; more than 97% of Hodgkin’s lymphoma patients present with a lesion above the diaphragm.23
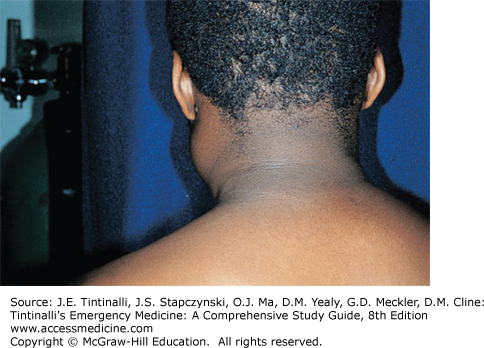
The differential diagnosis of cervical lymphadenopathy is extensive. See chapter 122, “Neck Masses in Infants and Children.” Diagnosis is confirmed by lymph node biopsy.
ED treatment before confirmatory diagnosis is usually directed at acute complications such as superior vena cava syndrome or upper airway compression (see “Complications of Pediatric Cancer and Oncologic Emergencies” section, below). Use caution with sedation, given the risk of airway compromise with mediastinal involvement. The need for admission is individualized. Do not give steroids to patients with significant lymphadenopathy if lymphoma is in the differential diagnosis. Following risk categorization, therapeutic management of Hodgkin’s lymphoma incorporates chemotherapy and radiotherapy. Significant toxicities, including cardiac, pulmonary, thyroid, and second malignant tumors in long-term survivors, prompted modification of existing protocols to decrease the radiation and chemotherapy exposure while maintaining good outcomes.25,26,27
Non-Hodgkin’s lymphoma accounts for 7% of cancer in children and adolescents in the United States, with little variation by age.28 Non-Hodgkin’s lymphoma includes a heterogeneous group of malignant neoplasms that can originate not only in the lymphatic system but in almost any organ in the body, including the skin, cortical bone, GI tract, and CNS. There is a male predominance,1 and the majority of cases have no known cause. Immunodeficiencies and previous exposure to immunosuppressive agents are known risk factors. In equatorial Africa, non-Hodgkin’s lymphoma accounts for almost 50% of childhood cancers and is almost universally associated with EBV.29 That association exists to a much lesser degree in developed countries.
The clinical presentation depends on the site and extent of disease. Constitutional symptoms are not common. Lymphadenopathy, a mass in virtually any location, hepatosplenomegaly, and cytopenias can all be seen. GI manifestations occur with abdominal tumors (usually Burkitt’s lymphoma). Mediastinal involvement may lead to pleural or pericardial effusions, upper airway obstruction from mass effect, respiratory symptoms, or superior vena cava syndrome (see “Complications of Pediatric Cancer and Oncologic Emergencies” section, below). Testicular, skin, and CNS or spinal manifestations are seen, but less commonly. The differential diagnosis is broad and includes infectious (tuberculosis, toxoplasmosis, EBV, Bartonella henselae, human immunodeficiency virus) and oncologic (Hodgkin’s lymphoma, leukemia, rhabdomyosarcoma) processes. Diagnosis is by biopsy.
Initial workup should include basic laboratory studies and a chest x-ray to evaluate for mediastinal disease. If there is concern for intra-abdominal obstruction (bowel or ureteral), ultrasonography or other imaging (e.g., abdominal and pelvic CT) may be indicated. CT can also further evaluate for chest mass or superior vena cava syndrome. Treatment is multiagent chemotherapy.
CNS tumors are the second most common pediatric cancer and the most common solid tumor in childhood, accounting for approximately 21% of all pediatric cancers.4 They are also the leading cause of cancer-related death in children.30 Survival is worse for infants and young children.31 For the emergency physician, the specifics of the tumor type are less important than tumor location and clinical effects.
The three most common categories of pediatric CNS tumors are astrocytomas, medulloblastomas, and ependymomas. For most, the cause is unknown.32 Specific genetic syndromes, including neurofibromatosis, tuberous sclerosis, and Li-Fraumeni syndrome, are predisposing factors. CNS tumors can also be secondary to high-dose cranial radiation for a previously treated childhood cancer.
There is heterogeneity in the presentation of CNS neoplasms depending on the location and extent of the tumor and the age of the child. Tumors can be infratentorial or supratentorial but are more often infratentorial in children. Symptoms are often nonspecific (headache, irritability, emesis, behavioral changes), which can delay diagnosis.
The classic presentation of a posterior fossa tumor or other tumors causing obstructive hydrocephalus is early morning headache and subsequent vomiting. These symptoms are believed to be due to a rise in intracranial pressure during sleep caused by hypoventilation (hypercarbia causes increased cerebral blood flow) and increased cerebral blood volume in the recumbent position. Although patients with brain tumors commonly have headaches, children with headaches are not statistically likely to have a brain tumor.33 Additional clinical signs such as a bulging fontanelle, rapidly increasing head circumference in an infant (from hydrocephalus), sunsetting (preferential downward gaze due to obstructive dilatation of the third ventricle with resultant tectal pressure and paresis of upward gaze), cranial nerve palsies (most commonly the sixth nerve), papilledema, or somnolence also suggest increased intracranial pressure.34 Infiltration of the brainstem can produce cranial nerve deficits, long tract corticospinal motor weakness, and cerebellar ataxia.
Symptoms of supratentorial neoplasms are dictated by location and include headache, endocrinopathies, declines in school performance, personality changes, motor weakness, and seizures. Craniopharyngiomas arise in the sellar region and produce visual changes due to their proximity to the optic chiasm, as well as significant endocrinologic abnormalities from hypothalamic dysfunction (including diabetes insipidus, stunted growth and sexual development, and hypothyroidism).
Most CNS malignancies are identifiable on CT, but MRI provides superior visualization of the tumor and the posterior fossa. The most serious threat is increased intracranial pressure leading to herniation. The treatment of increased intracranial pressure is addressed below in “Complications of Pediatric Cancer and Oncologic Emergencies.” Treat acute seizures. Give IV dexamethasone to treat vasogenic edema surrounding the tumor (see “Increased Intracranial Pressure” section, below). Consult with neurosurgery and pediatric oncology to arrange definitive treatment.
Neuroblastoma is a malignant tumor that arises from primitive ganglion cells of the sympathetic nervous system. It accounts for 7% of all childhood cancers and is the most common solid extracranial tumor of childhood and the most common neoplasm in the first year of life.4,35
The clinical features are highly variable and depend on tumor size and location. Neuroblastoma may arise anywhere along the sympathetic nervous system; the adrenal gland is the most common primary site, but it may also arise from other intra-abdominal sites, the chest, and the neck. Metastasis is quite common (through circulatory or lymphatic channels) to adjacent lymph nodes, liver, skin, bone, and bone marrow.
The variability in tumor location and extent explains the array of signs and symptoms that may lead to the diagnosis of neuroblastoma. One presentation is a painless abdominal mass. Mass effect from the tumor can lead to compression of the bowel, bladder, venous, or lymphatic structures. Thoracic masses can also lead to compression, causing respiratory distress or superior vena cava syndrome. Paravertebral tumors can invade the spinal canal, causing spinal cord compression. Horner’s syndrome (ptosis, miosis, and anhidrosis) may result from disease involving the superior cervical ganglion.
Unique presentations of neuroblastoma that require a high index of suspicion include proptosis and periorbital ecchymoses (“raccoon eyes”) or swelling, characteristic of orbital neuroblastoma metastases. Neuroblastoma is the most common primary childhood cancer to metastasize to the orbits, and orbital findings can be the primary manifestation of the tumor.36 Opsoclonus-myoclonus is a paraneoplastic syndrome also highly associated with occult neuroblastoma.
Obtain basic laboratory studies. Cytopenias suggest bone marrow involvement. X-rays, US, or CT in the ED may detect an intra-abdominal or thoracic mass. Because neuroblastomas often lead to increased levels of catecholamines, the metabolites of which are detectable in the urine, urine homovanillic acid and vanillylmandelic acid can aid in diagnosis. Bone marrow and tumor biopsies confirm the diagnosis.
Treatment includes surgical resection and/or radiation therapy and chemotherapy. Some undergo stem cell transplants.
Wilms’ tumor, or nephroblastoma, is a malignant embryonal renal tumor that affects children predominantly under the age of 5 years.4 Overall survival rates are excellent, at approximately 90%.37
The classic presentation of Wilms’ tumor is an asymptomatic abdominal mass, sometimes noted while dressing or bathing the child. Vigorous or excessive palpation of the mass can cause tumor rupture. As opposed to neuroblastoma (given that the age group and abdominal mass at the time of presentation can be similar), children often appear remarkably well with no systemic symptoms. Metastases at the time of diagnosis are uncommon, but can occur to the lungs, liver, or lymph nodes. Hypertension due to increased renin production is present in 25% of cases.38 Less common symptoms include anorexia, weight loss, emesis, hematuria, or abdominal discomfort. There can be significant mass effect from the tumor, leading to respiratory distress or intra-abdominal obstruction.
The differential diagnosis includes benign hydronephrosis, neuroblastoma, hepatoblastoma, lymphoma, sarcoma, and germ cell tumors.39 Ultrasonography is appropriate for initial abdominal imaging in the ED. Obtain a chest x-ray to identify pulmonary metastases (Figure 143-2). Obtain basic laboratory studies, urine catecholamines to rule out neuroblastoma, and urinalysis. CT or MRI identifies the extent of the disease. Definitive treatment is surgical resection and chemotherapy, with or without radiation therapy.
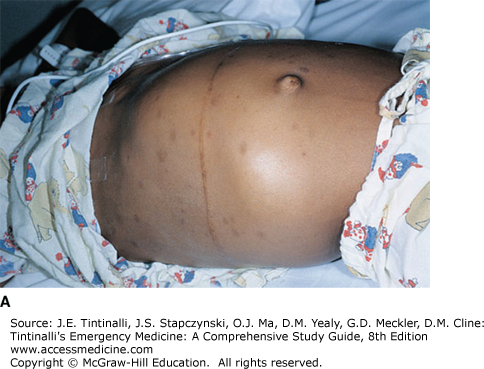
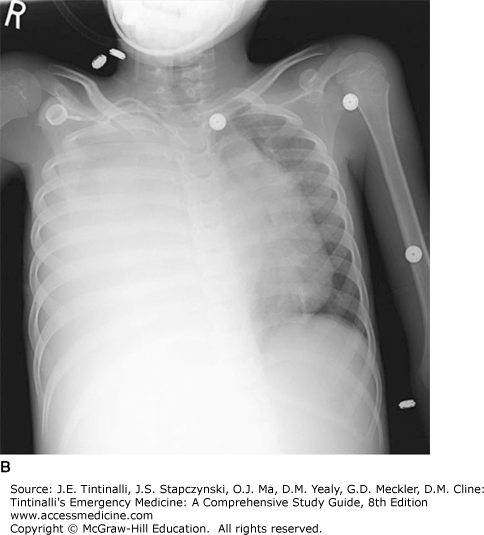
FIGURE 143-2.
Wilms’ tumor. A. A 7-year-old child presented with a huge abdominal mass and respiratory distress. Pelvic CT scan followed by laparotomy confirmed the mass as Wilms’ tumor. Because of the retroperitoneal location of the kidney, these tumors can be quite large at diagnosis without significant impingement of other vital structures. B. Right-sided pleural effusion due to metastatic disease. Almost complete opacification of the right hemithorax with mediastinal shift to the left. [Reproduced with permission from Shah BR, Lucchesi M: Atlas of Pediatric Emergency Medicine, © 2006, McGraw-Hill, New York.]
Retinoblastoma is the most common pediatric intraocular malignancy.36 Ninety percent of cases are diagnosed in children younger than 3 years of age.40 There are both heritable and nonheritable forms, and the majority of cases of nonheritable disease are unilateral. The retinoblastoma (RB) gene was the first tumor suppressor gene discovered in the human genome.41 Heritable forms are usually due to a germline mutation in the RB1 gene and present within the first year of life with bilateral disease.
Leukocoria (“white pupil”), in which the white light reflects off of the tumor instead of red light reflecting off of the retina, is the most common presenting sign (Figure 143-3). This may be noted by a primary physician or occasionally by family members when they note the leukocoria in photographs. Other signs may include strabismus, decreased visual acuity, redness, pain, glaucoma, and proptosis (late stage). Young children do not often report visual complaints. The retinoblastoma can spread into the surrounding orbit, as well as metastasize into the CNS and viscera.36
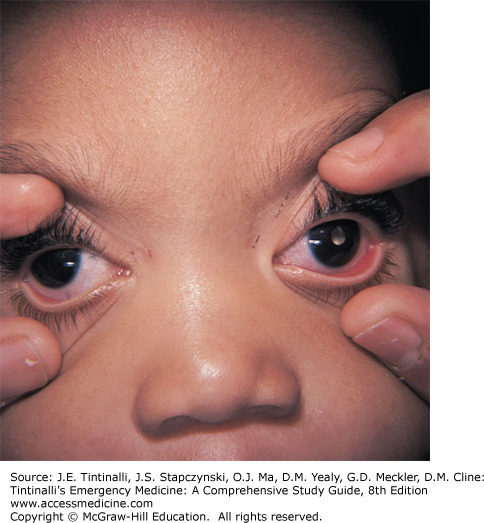
If leukocoria or other signs suggestive of an intraocular tumor are noted, consult ophthalmology in the ED. Definitive diagnosis is usually obtained by exam and imaging under anesthesia. Obtain a CT or MRI if there is evidence of orbital inflammation. Treatment options depend on the extent of the disease and include enucleation, chemotherapy, cryotherapy, brachytherapy, thermotherapy, radiotherapy, and laser photocoagulation.42
Germ cell tumors are a heterogenous group of neoplasms that can develop in males and females at any age, although they are more common in adolescents and young adults.4,43 Extragonadal tumor locations, including sacrococcygeal, mediastinal, and intracranial, are more common in young children, whereas during and after puberty, the primary location is the gonads.43
Testicular germ cell tumors are the most common solid tumor in young adult men (Figure 143-4).44 An undescended testicle increases the risk for testicular cancer 10- to 50-fold.45 Prognosis for these tumors is extremely good, even if metastases are present, with an overall cure rate of 85% to 90%.44 Despite that fact, they remain a significant cause of death in young males. This highlights the importance of encouraging young male patients to perform testicular self-examinations. The median age of diagnosis for ovarian germ cell tumors is 16 to 20 years.46 Due to the intra-abdominal location, ovarian tumors may go undetected for a longer amount of time than testicular tumors; this increases the potential for rupture.47
FIGURE 143-4.
Testicular tumor. This patient presented with a painless left testicular mass highly suspicious for cancer. [Photo contributed by Patrick McKenna, MD. Reproduced with permission from Knoop K, Stack L, Storrow A, Thurman RJ: Atlas of Emergency Medicine, 3rd ed. © 2010, McGraw-Hill, New York.]
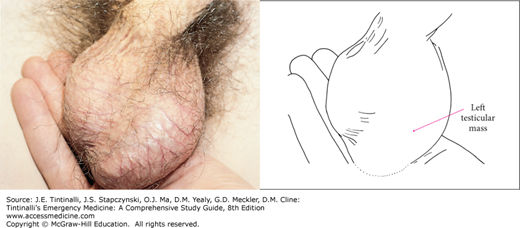
Testicular tumors typically present with an asymptomatic, nontender testicular mass. Ovarian tumors are most likely to present with abdominal pain (85%).46 Other ovarian symptoms include abdominal distention, vaginal bleeding, and weight gain. Ten percent of patients with ovarian tumors presenting with abdominal pain from tumor rupture, hemorrhage, or torsion are misdiagnosed.46 Tumors can metastasize, most commonly to the lymph nodes, liver, lungs, and CNS.
A testicular mass or abdominal mass may be palpated upon exam. Ultrasonography identifies an ovarian or scrotal mass. CT delineates the extent of disease. Serum tumor markers can be added to the basic laboratory tests. Treatment includes resection, chemotherapy, and/or radiotherapy. Given that many patients are of reproductive age, treatment attempts to preserve fertility if possible.
The term sarcoma refers to a diverse group of malignant neoplasms derived from mesenchymal cell origin that can arise from virtually any location at any age. Sarcomas are divided into two main types: soft tissue sarcomas and bone sarcomas. There are over 70 recognized subtypes. The most common subtypes diagnosed in pediatrics are rhabdomyosarcoma, osteosarcoma, and Ewing’s sarcoma.
Rhabdomyosarcoma accounts for 3% of childhood cancers and 2% of adolescent cancers.4 Risk factors include a number of inherited cancer predisposition syndromes (e.g., Li-Fraumeni, neurofibromatosis type I). The embryonal subtype (>75% of cases) is most common in children <5 years old, tends to occur in the head and neck, and has a better prognosis. The alveolar subtype can occur at any age, is more aggressive, and is commonly found in the trunk or extremities. Orbital rhabdomyosarcoma accounts for 10% of all rhabdomyosarcoma cases.36
Signs and symptoms depend on location, but a painless mass is characteristic. Evaluation in the ED may begin with ultrasonography. More extensive evaluation steps are determined by the oncologist. Treatment includes a combination of surgical excision, chemotherapy, and radiotherapy.
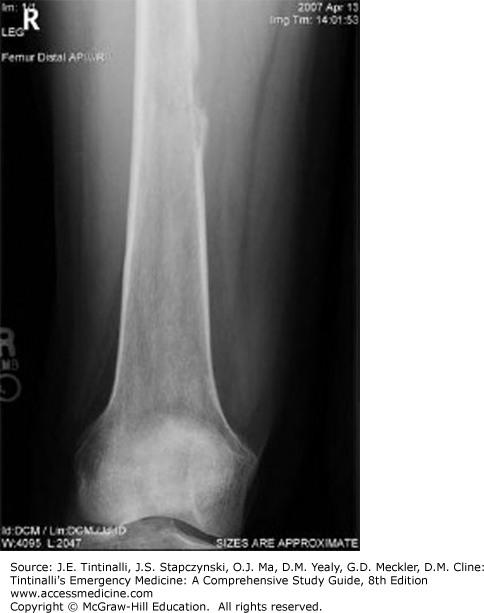
Osteosarcoma, also called osteogenic sarcoma, is the most common primary pediatric bone tumor and is among the most frequent causes of cancer-related death.48 Incidence peaks in adolescence, particularly during a growth spurt. It can occur in any bone, but the majority of cases arise from the metaphyses of long bones. Over half of cases originate near the knee joint, either at the distal femur or at the proximal tibia.49 Twenty percent of patients have metastases at the time of diagnosis, most commonly in the lungs, followed by bone.4 Previous radiation during treatment for a different pediatric neoplasm increases the risk of osteosarcoma, as do several genetic syndromes.
Nonspecific symptoms can lead to a delay in diagnosis. The most common presentation is an adolescent with persistent bone pain that worsens at night or with activity. Soft tissue swelling may be noted on exam.
Diagnosis is suggested by plain radiography (Figure 143-5). The radiograph may demonstrate a lytic lesion with cortical destruction near the metaphysis. The classic radiographic finding is the “sunburst” appearance of periosteal reaction. Pathologic fracture is an uncommon presentation.50 The differential diagnosis of radiographically similar lesions includes Ewing’s sarcoma as well as more benign lesions. Diagnosis is by biopsy, and treatment includes chemotherapy and limb-sparing surgery. Osteosarcoma is radiation resistant.
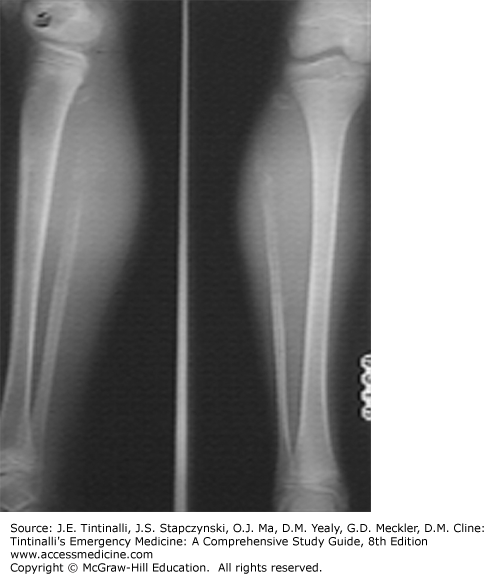
Ewing’s sarcoma is an aggressive tumor of uncertain origin that can occur in the bone or soft tissues. It is more common in older children and adolescents, but the incidence is half that of osteogenic sarcoma.51 Hematogenously spread metastases are present in approximately 25% of patients at the time of diagnosis and are most commonly located in the lungs, bone, and bone marrow.4 Metastatic disease is the most significant prognostic factor.52
Ewing’s sarcoma typically presents with persistent pain at the tumor site. Tenderness or swelling may be noted on physical exam. The long bones (femur, tibia, humerus) and the axial skeleton (pelvis, ribs, spine) are the most common sites of disease.
Plain radiographs of the primary tumor site may reveal the characteristic “moth-eaten appearance” of the destructive lesion, or the “onion peel” appearance of the periosteal reaction (Figure 143-6). Diagnosis is by biopsy, and treatment is multidisciplinary, including chemotherapy and radiotherapy or surgery.
COMPLICATIONS OF PEDIATRIC CANCER AND ONCOLOGIC EMERGENCIES
For the child with cancer, the road to recovery is inevitably interrupted by complications. These complications can be generally classified as infectious (e.g., fever and neutropenia), metabolic (including tumor lysis syndrome, hypercalcemia, and the syndrome of inappropriate antidiuretic hormone secretion), and structural (superior vena cava syndrome, spinal cord compression, and increased intracranial pressure).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


