ODOR: UNUSUAL
ALISON ST. GERMAINE BRENT, MD
The human nose is able to discriminate approximately 4,000 odors! Occasionally, parents bring an infant or a child to the emergency department (ED) complaining of an unusual smell. Adolescents are more likely to note a new or an unusual odor themselves and present to the ED with specific complaints.
Olfaction is not a sense that most medical providers are trained to use, quantify, or describe. Before the development of sophisticated laboratory tests, clinicians relied heavily on the sense of smell and often made clinically perceptive diagnoses by aroma alone. Today, by incorporating the sense of smell into a clinical skill set, an astute provider can make a presumptive diagnosis of a metabolic disorder, intoxication, or infection and institute lifesaving therapy prior to laboratory confirmation.
PATHOPHYSIOLOGY
The olfactory area extends from the roof of the nasal cavity approximately 10 mm down the septum and superior turbinates bilaterally. The exact mechanism of stimulation of the olfactory receptors is unknown. Smell is more acute in the darkness and is believed to be linked to blood cortisol levels.
The unique odor emitted by a person is produced by a combination of body secretions and excretions, particularly those from the oropharynx, nasopharynx, and the respiratory tract, plus aromas from the skin and cutaneous lesions, urine, feces, and flatus. The most significant components of odor in healthy humans are the apocrine glands. These secretions are initially odorless, but bacterial breakdown that results in fatty acid production can cause an offensive odor. Body odor is altered by hygiene, metabolism, toxins, infections, and systemic disease.
When a child is unable to detect odor, anosmia should be considered. When a child complains of strange odors, especially if no one else is able to identify them, temporal lobe epilepsy should be contemplated.
DIFFERENTIAL DIAGNOSIS
A number of conditions, including metabolic disorders, intoxications, infections, dermatologic conditions, foreign bodies, various abnormalities of the body orifices, and a variety of systemic diseases, may result in noxious odor (Table 45.1).
Metabolic Disorders
The most common metabolic disorder with a characteristic odor is diabetic ketoacidosis (Table 45.2). The characteristic breath odor is described as sweet, fruity, or similar to nail polish remover. This distinctive aroma is caused by acetone from the breakdown of ketones bodies to ketoacids (acetone, acetoacetate, and betahydroxybutirate). It is important to note that any condition that results in a marked metabolic acidosis with ketosis will result in the characteristic sweet or fruity breath.
Inborn errors of metabolism that result in altered body, breath, or skin odors are unusual individually, but as a composite, they reflect a significant percentage of life-threatening illnesses of infancy (see Chapter 103 Metabolic Emergencies). Although definitive diagnosis depends on specific identification of serum and urine amino and organic acid levels, many such conditions are associated with a positive urine ferric chloride test, which when performed in the ED can yield presumptive diagnosis (Table 45.2).
Phenylketonuria is a disorder of amino acid metabolism associated with a deficiency of phenylalanine dehydroxylase and dihydropteridine reductase, which forces use of minor metabolic pathways of phenylalanine, resulting in the buildup of phenylacetic acid. It is the buildup of phenylacetic acid in sweat and urine that causes the characteristic aroma described as musty, mousy, horsey, wolf-like, barny, or stale sweaty locker-room towels. Clinical features of untreated phenylketonuria include white-blond hair, blue eyes, fair complexion, eczema, microcephaly, hypertonicity, seizures, and progressive mental deterioration. Although neonatal screening detects most of these cases, the observation of a characteristic odor in an infant should prompt immediate appropriate laboratory studies, which may include a urine ferric chloride test in the ED. Prompt diagnosis and dietary restriction of phenylalanine promote normal development.
Maple syrup urine disease is caused by a metabolic defect in the decarboxylation of the ketoacids of the branch-chain amino acids (leucine, isoleucine, and valine), which results in their accumulation in the blood. Sotolon (4,5-dimethyl-3-hydroxy-2[5H]-furanone), a metabolite of isoleucine in the urine, produces the characteristic odor described as maple syrup, caramel-like, malty, or boiled Chinese herbal medicine. Children with this disorder can have variable clinical manifestations, ranging from decreased appetite, vomiting, and ataxia, to progressive acidosis, seizures, coma, and death. Prompt diagnosis and limitation of dietary branched-chain amino acids promotes normal development.
Oasthouse urine disease, or methionine malabsorption syndrome, is caused by defective transport by the intestines and kidneys of methionine and, to a lesser extent, leucine, isoleucine, valine, tyrosine, and phenylalanine. The unabsorbed methionine in the gut is broken down by colonic bacteria to α-hydroxybutyric acid, which causes the distinctive odor, described as dried malt or hops (as in breweries), dried celery, or yeast. Clinical presentation includes fair hair and skin, hyperpnea, extensor spasms, fever, edema, and mental retardation. Successful treatment consists of a methionine-restricted diet.
The odor of sweaty feet syndrome, or isovaleric acidemia, is caused by a defect in the catabolism of leucine. The characteristic odor described as sweaty feet or socks or ripe cheese comes from the buildup of isovaleric acid. Clinically, children experience vomiting, dehydration, acidosis, and slowly progressive mental deterioration. Treatment includes restriction of leucine in the diet.
TABLE 45.1
CLINICAL SOURCE AND CAUSE OF UNUSUAL ODORS
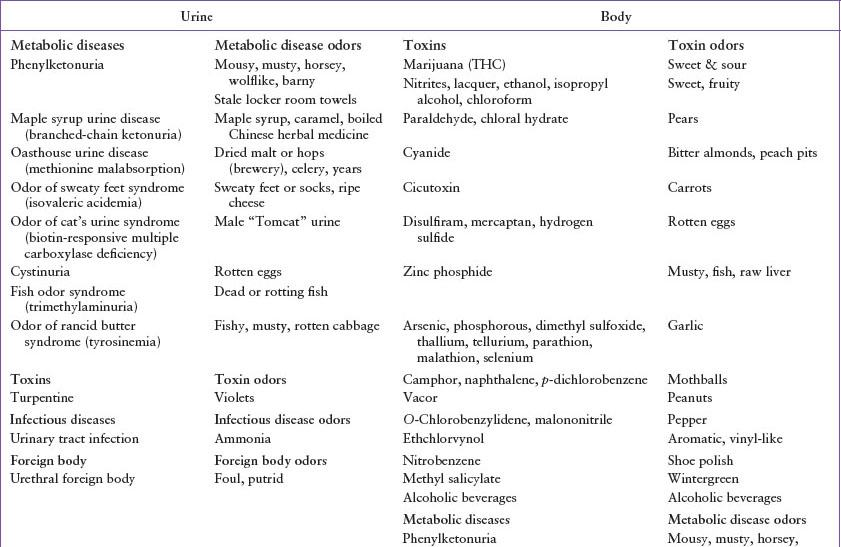



In the odor of cat’s urine syndrome, the enzymatic defects are in the biotin-dependent enzymes β-methylcrotonyl-CoA carboxylase, pyruvate carboxylase, and propionyl-CoA carboxylase. The cause of the distinctive aroma of cat urine, referred to as male or “tomcat” urine, is unknown. Clinically, children have failure to thrive, ketoacidosis, and neurologic symptoms similar to Werdnig–Hoffmann disease. Treatment consists of a low-leucine diet and the addition of biotin.
TABLE 45.2
METABOLIC DISEASE ASSOCIATED WITH UNUSUAL ODORS
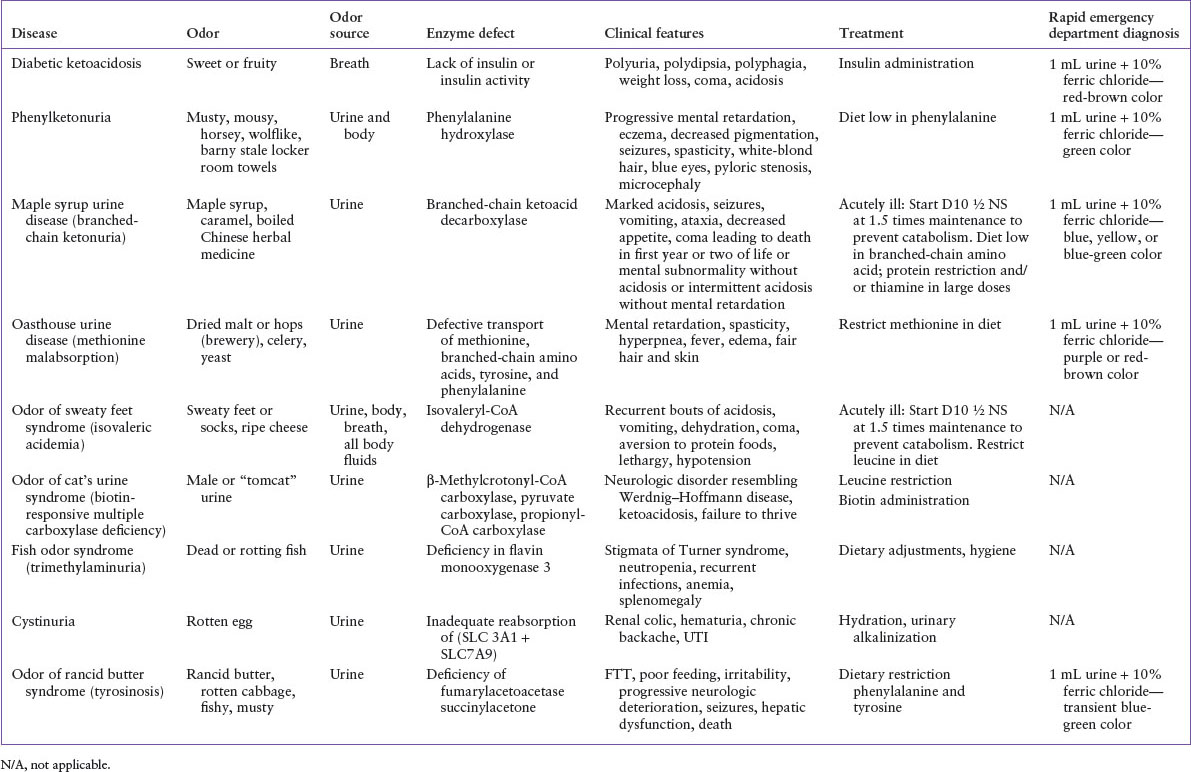
Fish odor syndrome, trimethylaminuria, results in the unmistakable odor of dead or rotting fish in the urine, breath, and sweat. This aroma is produced by a buildup of trimethylamine, caused by an inherited deficiency in flavin monooxygenase 3, the vital enzyme for the metabolism of trimethylamine. Clinical and laboratory presentation includes stigmata of Turner syndrome, history of recurrent pulmonary infections, a normal complement of chromosomes, neutropenia, and abnormal platelet function. While the disease itself is not life-threatening, it can be devastating from a psychosocial perspective and has been associated with suicide attempts, hence practitioners need to remain attentive to this often overlooked diagnosis. Unfortunately there is no cure or dietary adjustments that can reduce the excretion of trimethylamine and attention to body hygiene may be helpful adjuncts.
Tyrosinemia, the odor of rancid butter syndrome, is identified by a distinctive cabbage-like odor. This syndrome is caused by a deficiency of fumarylacetoacetase which results in the buildup of succinylacetone, the decarboxylation product of succinyl acetoacetate, a compound derived from the tyrosine catabolic intermediate fumarylacetoacetate. Clinical presentation may include failure to thrive, which may precede the appearance of more dramatic findings in tyrosinemia, including vomiting and diarrhea, which rapidly progressto bloody stool, lethargy, and jaundice, irritability, seizures, coma, progressive neurologic deterioration, and can result in early death secondary to infection and liver failure. In some cases, restriction of dietary phenylalanine and tyrosine has been helpful.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


