Remimazolam
Dexmedetomidine
Etomidate analogsa
Fospropofol
Class
Benzodiazepine
α2 adrenoceptor agonist
Imidazole
Propofol prodrug
Receptor
GABAA
α2 adrenoceptors
GABAA
GABAA
Sedative dose
0.1–0.2 mg/kg
1 mcg/kg over 10 min, then 0.2–0.7 mcg/kg/h infusion
To be determined
6.5 mg/kg followed by 1.6 mg/kg at 4 min intervals
Dose reduction for elderly
No
Yes
To be determined
Yes
Dose reduction for hepatic impairment
No
Yes
To be determined
Limited data but dose reduction advised
Dose reduction for renal impairment
No
No
To be determined- carboxylic acid metabolite may accumulate in renal impairment
No dose reduction with creatinine clearance > 30 ml/min
Limited data for patients with creatinine clearance < 30 ml/min
Onset
1.5–2.5 min
5–8 min
To be determined- rapid in animal models
4–8 min
Metabolism
Ester hydrolysis
Hepatic metabolism via cytochrome p450 and glucuronidation
Ester hydrolysis
Fospropofol metabolized to propofol by endothelial and hepatic alkaline phosphatases
Propofol metabolized by hepatic and erythrocyte dehydrogenases
Offset
6.8–9.9 min
6 min
To be determined- rapid in animal models
5–18 min
Active metabolite
No
No
Carboxylic acid metabolite
No
Benzodiazepine use for procedural sedation has two main limitations: the absence of analgesic properties, and persistence of sedative effects beyond the duration of the procedure. This latter feature relates to both diffuse drug distribution and prolonged elimination. The use of midazolam, which has the shortest half-life of any of the benzodiazepines, can lead to prolonged sedation and unpredictable recovery due to a half-life of 1.8–6.4 h and accumulation of an active metabolite, α-hydroxy-midazolam, which has a sedative effect [4, 5].
Remimazolam, in contrast, may bypass these limitations of benzodiazepines, and may indeed have many of the properties of the ideal sedative agent for use in the out-of-OR setting [6, 7]. In addition to producing dose-dependant sedation, remimazolam has a rapid offset when compared to midazolam. This is due to metabolism and clearance by tissue esterases, in addition to an inactive metabolite [8]. Another important feature of remimazolam for the out-of-OR setting is the lack of accumulation, with the context-sensitive half-time similar to that of remifentanil [9].
Uses
Pre-operative | Pre-procedural premedication |
Intra-operative | Initiation and maintenance of procedural sedation |
Rapid onset and offset sedation for procedural sedation [10] | |
Co-induction of anaesthesia | |
Benzodiazepine anaesthesia | |
Post-operative | Shorter duration of recovery from anesthetic/sedation, owing to reduction in intraoperative anesthetic andopioid requirements |
Structure
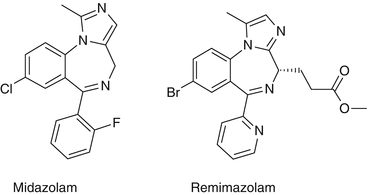
The structures of midazolam and remimazolam are presented in Fig. 31.1. The significant difference between the two molecules is the introduction of a carboxylic ester linkage to remimazolam, which allows metabolism by non-specific tissue esterases in the blood- in a similar way to remifentanil.
Dosing
Sedative dose 0.1–0.2 mg/kg [10]
Due to organ-independent elimination, no dosage adjustments are required for patients with hepatic or renal impairment.
Age-related deterioration of hepato-renal drug handling is unlikely to have impact on remimazolam’s metabolism
Kinetics
Like other benzodiazepines, remimazolam acts on GABA receptors, specifically GABAA [8], modulating the effects of GABA at the GABA receptors
Onset 1.5–2.5 min [10]
Metabolized by dose-independent ester hydrolysis into inactive metabolite CNS 7054
Rapid clearance 70.3 ± 13.9 L/h (midazolam clearance 23 ± 4.5 L/h) [8]
Volume of distribution 23 ± 4.5 L/h (midazolam volume of distribution 81.8 ± 27.1 L/h) [8]
Half life 0.75 ± 0.15 h [8]
Mean offset time 6.8–9.9 min [10]
Minimal accumulation- context sensitive halftime 7–8 min after a 2-h infusion [9]
Slow clearance of inactive metabolite (4.22 ± 1.25 L/h) with terminal half-life of 2.89 ± 0.65 h
Reversed by flumazenil
Adverse Effects
At the time of publication, there was ongoing recruitment for a phase III trial investigating efficacy and adverse effects of remimazolam in patients undergoing colonoscopy [12]. From the results of the phase I and II trials of remimazolam, the adverse effects of remimazolam appear to be similar to those of other benzodiazepines, including hypotension, respiratory depression, and desaturation [9, 10].
Dexmedetomidine
Clonidine was the first α2-adrenoceptor agonist, and was formulated in the 1960s. It was initially marketed as a nasal decongestant but was subsequently recognized as an effective antihypertensive and sedative drug. Dexmedetomidine, which is also an α2-agonist, was approved by the Food and Drug Administration at the end of 1999 for short-term (<24 h) analgesia and sedation in the intensive care unit. Its use has since expanded to include the perioperative period.
Dexmedetomidine has greater specificity for the α2-adrenoceptor compared to clonidine (ratios of α2:α1 activity, 1620:1 for dexmedetomidine, 220:1 for clonidine) [13]. Presynaptic α2- adrenoceptors regulate the release of norepinephrine and adenosine trisphosphate through a negative feedback mechanism, which results in the analgesic effects of dexmedetomidine (Fig. 31.2). The sedative effects of dexmedetomidine result from agonism of central α2 receptors in the presynaptic neurons and subsequent inhibition of neuronal firing in the brain, particularly the locus coeruleus, and in the spinal cord [14].
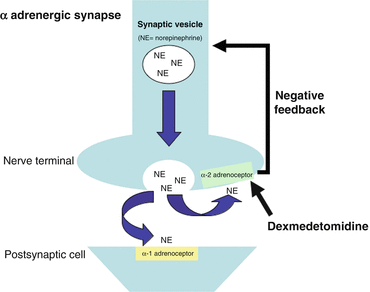
Fig. 31.2
The action of dexmedetomidine at a synapse
The analgesic effects of dexmedetomidine are a result of:
- 1.
direct α2-adrenoceptor agonism and inhibition of norepinephrine release
- 2.
α2-adrenoceptor modulation of G-protein-gated potassium channels, resulting in membrane hyperpolarization and decreased firing rate in excitable cells in the CNS
- 3.
G protein coupled reduction in calcium conductance through cell membranes and inhibition of neurotransmitter release [15]
There are many advantages to dexmedetomidine as an out-of-OR sedative. Because its actions are not mediated by the GABA-mimetic system, it has sedative, analgesia, and antishivering properties, but does not cause respiratory depression [16]. ‘Cooperative sedation’ is a term used to describe the sedative effect of dexmedetomidine, whereby the patient can be deeply sedated yet rousable and able to interact with healthcare providers [17].
Uses
Pre-operative | Pre-procedural premedication [18] |
Intra-operative | Initiation and maintenance of procedural sedation |
Dose-dependant sedation without respiratory depression [21] | |
Arousability maintained at deep levels of sedation [22] | |
Attenuation of sympathoadrenal effects of surgical stimulationand endotracheal intubation [23] | |
Post-operative | Reduced duration of recovery from anesthetic/sedation,owing to reduction in intraoperative anesthetic and opioidrequirements |
Reduction in analgesic requirements [24] | |
Other |
Structure
Dexmedetomidine is an imidazole compound and is the pharmacologically active dextroisomer of medetomidine, which displays selective α2-agonism.
Presentation
Dexmedetomidine is presented in 200 mcg/2 mL (100 mcg/mL) glass vial to be used after dilution. Dexmedetomidine must be diluted with 48 ml 0.9 % sodium chloride injection to achieve a concentration of 4 mcg/ml.
Dosing
Initial loading dose 1 mcg/kg over 10 min, followed by continuous IV infusion of 0.2–0.7 mcg/kg/h [27]
For patients >65 years a loading infusion of 0.5 mcg/kg over 10 min and a reduction in the maintenance infusion should be considered [27]
Hepatic impairment- dose reduction recommended [27]
Renal impairment- no dose reduction required [28]
Kinetics
Time to onset of action 5–8 min [27]
Peak effect 10–20 min [27]
Peak plasma concentrations achieved within 1 h after continuous infusion [27]
Distribution half-life (t1/2) of approximately 6 min; a terminal elimination half-life (t1/2) of approximately 2 h [27]
Highly bound to plasma proteins (94 %) [27]
Lipophilic- Steady-state volume of distribution (Vss) of approximately 118 l
Metabolism by the liver via cytochrome P450 & glucuronidation [13]
Clearance is approximately 39 L/h [27]
Mean offset time after single dose 6 min [27]
Context sensitive halftime increases, depending on duration of infusion- ranging from 4 min after a 10 min infusion to 250 min after an 8 h infusion [29]
No active metabolites
Inactive metabolites excreted in urine (~95 %) and faeces (4 %) [27]
Adverse Effects [27]
Cardiovascular
Hypotension incidence (56 %) and bradycardia incidence (42 %), both of which are due to inhibition of central sympathetic outflow [30, 31]
Hypertension on initial bolusing, owing to peripheral α2B-adrenoceptor stimulation of vascular smooth muscle incidence (16 %) [30, 31]
Arrhythmias including atrial fibrillation (4 %), extrasystoles, supraventricular tachycardia, ventricular arrhythmia and heart block
Respiratory
Respiratory depression incidence (37 %)
Respiratory failure incidence (6 %)
Acute respiratory distress syndrome incidence (3 %)
Pleural effusion incidence (2 %)
CNS
Agitation incidence (8 %)
Anxiety incidence (5 %)
GI
Nausea incidence (11 %)
Constipation incidence (6 %)
Dry mouth incidence (4 %)
Others
Hypokalaemia incidence (9 %)
Pyrexia incidence (7 %)
Hyperglycaemia incidence (7 %)
Anemia incidence (3 %)
Oliguria incidence (2 %)
Etomidate Analogs- MOC-Etomidate & Carboetomidate
Etomidate is a rapidly acting imidazole-based intravenous sedative-hypnotic agent that is used to induce general anesthesia through potentiation of GABAA receptor activation. As is the case with other intravenous anaesthetic agents, etomidate’s hypnotic action ceases following bolus delivery when it is redistributed from the brain to other tissues. Etomidate undergoes elimination by the liver with a half-life of several hours [32]. One of the main advantages of etomidate over other induction agents is its ability to maintain hemodynamic stability even in the setting of cardiovascular compromise [33]. The predominant issue surrounding the use of etomidate however, is adrenocortical suppression [34]. Etomidate potently inhibits 11β-hydroxylase, an enzyme in the biosynthetic pathway leading to adrenocortical steroid synthesis [35, 36]. Alternative etomidate analogs are currently under investigation in the hopes of developing a rapidly metabolized hypnotic agent with the haemodynamic stability of etomidate but without the adrenocortical suppression characteristic of the parent compound. At the time of publication, these agents were under development and had not yet undergone human testing.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


