Water soluble
Stable in solution
Long shelf life
No pain on intravenous injection
Nonirritant if given subcutaneously
Painful on arterial injection
No sequelae after intra-arterial injection
Low incidence of venous sequelae (thrombophlebitis)
Small injection volume
Lack of hypersensitivity reactions
Lack of intravascular hemolysis
Does not necessarily need to have analgesic properties
No induction complications such as excitatory effects, tremor, spontaneous movements, or hypertonus
No muscular reaction as maintenance of jaw tone may facilitate airway maintenance
No effect on uterine tone
Does not induce episode of malignant hyperpyrexia nor induce acute porphyria
Ideally free of fetal teratogenicity
The present use of modern intravenous agents to provide an alternate to the volatile agents for the maintenance of anesthesia has stemmed from the development of four intravenous agents which fulfilled most of these properties (ketamine, Althesin (alfaxalone -alfadolone acetate), etomidate, and propofol ), as well as showing the important pharmacokinetic-pharmacodynamic features needed. The latter features ideally include being water soluble and hence no likelihood of solvent toxicity; rapid blood-brain equilibration, high potency, and a steep dose-response curve to enhance titratability; rapid plasma clearance and small volume of distribution to minimize the potential for tissue accumulation; and the absence of active metabolites. Other desirable features are minimal cardiovascular and respiratory depression and low potential for histamine release.
Of those drugs listed above, propofol is considered by many present-day observers to be near to the ideal. However, time and experience have shown its use to be associated with a number of important issues and side effects. Because of its water insolubility, the drug must be formulated in a lipid solution, with the added issue of the risk of bacterial contamination if vials are left open to the air. Early on in the evaluation of the agent, there was noted to be significant pain on intravenous injection, especially when administered into small veins on the back of the hand. Although all intravenous agents have cardiovascular effects, the dose-related cardiovascular and respiratory depression seen with propofol , especially when the drug is given in conjunction with opioid drugs, was greater than with the other agents. Propofol also has a liability to development of the propofol infusion syndrome (PRIS) , with hyperlipidemia and metabolic acidosis, when given for prolonged sedation especially in the intensive care unit [2]. Other issues that have been researched include the stability of the solvent and the need for antimicrobials to prevent bacterial contamination.
Because of the drawbacks associated with the use of the original 10 % soy bean oil emulsion for the formulation of propofol , there have been a number of different solutions investigated—these include solutions with a lower quantity of soy bean oil, the use of solutions containing both short-chain and long-chain fatty acids, the use of albumen emulsions, the use of cyclodextrins, and the development of aqueous formulations by presentation of the drug as a phosphate or other salt.
The observation of these side and untoward effects associated with the use of propofol has led to considerable effort over the last decade or so by both the pharmaceutical industry and other researchers to develop either new formulations of existing agents or new chemical entities able to provide both induction and maintenance of anesthesia.
These researches have been addressed in this chapter by considering the researches to improve the profile of the archetypal IV agent propofol and then to consider other new chemical entities for the induction and maintenance of anesthesia. Discussion of these has been divided into those that have undergone or are undergoing clinical evaluation and other drugs presently at the preclinical phase.
New Propofol Formulations
The aim for any new propofol formulation must be to show an improved pharmacologic profile compared with the original “Diprivan” formulation. To do this, there must be a reduced incidence of pain on injection and an improved profile of the other minor untoward side effects. But reformulation must not cause any significant change in the drug’s PK-PD profile. If the incidence of PRIS is to be reduced, a non-lipid formulation should ideally be developed. It will also be desirable that any new formulation achieved a faster onset of effect. The one other key adverse effect is the risk of bacterial contamination; however, support of bacterial growth can be obtunded by addition of benzyl alcohol, sulfite, or EDTA.
The problem of pain on injection is more debatable as the lipid solvent is not associated with pain when given alone or when used to solubilize other agents [3], suggesting that the pain may be due to the propofol itself. New formulations have focused either on the choice of lipid or other solvent used to solubilize propofol or on the formulation of water-soluble preparations of the drug. The Lipuro formulation of propofol is solubilized in medium-chain triglycerides rather than long-chain triglycerides. It has an identical pharmacokinetic-pharmacodynamic profile to the “Diprivan” formulation but is associated with less pain on injection. However, this formulation is not available in the USA because of the lack of EDTA in the solvent. Propofol emulsions contain oil droplet average sizes of 0.15–0.5 μm (fine macroemulsions) [4]. If the droplet size is less than 0.1 um, the emulsion is known as a microemulsion ; these are more highly stable in solution because of a larger droplet size, but they require the addition of surfactants to retain the drug in solution [5]. Examples of these microemulsions include Aquafol (Daewon Pharmaceutical Co., Ltd, Seoul, Korea) which contains 1 % propofol , 8 % polyethylene glycol 600 hydroxystearate (solutol HS 15; BASP Co. Ltd., Seoul, Korea) as a nonionic surfactant, and 5 % Glycofurol (Roche, Basel, Switzerland) as a cosurfactant. Its use has been compared with Diprivan 1 % in volunteers with similar pharmacokinetic and pharmacodynamic properties and safety profiles [5]. However the doses of surfactant and cosurfactant used in this formulation limited its administration to a maximum of 100 ml Aquafol per day.
This has led to the microemulsion being reformulated with another, better tolerated cosurfactant (Purified poloxamer 188; PP188). When investigated in the rat, the kinetics of this formulation were found to be nonlinear [6]. There are no studies to date in man which report on the incidence or not of pain on injection to this formulation. It has been suggested that this might increase with microemulsion formulations due to a greater amount of free propofol in the aqueous phase [7].
Three aqueous formulations of propofol solubilized using cyclodextrins have been studied in various animal species [8]. These use hydroxypropyl-gamma-cyclodextrin, hydroxypropyl-β-cyclodextrin (HP-βCD), or sulfobutyl-β-cyclodextrin as the solvent. The hydroxypropyl-β-cyclodextrin formulation showed favorable physicochemical and biological properties as a water-based formulation. Early porcine studies show an equivalent PK-PD profile to “Diprivan.” Again, there are no reported studies in patients. However, when formulations were used containing large amounts of a cyclodextrin complexing agent (20 % w/v), IV dosing of propofol -hydroxypropyl-β-cyclodextrin in rats caused immediate bradycardia of variable duration, while the solvent alone had no effect [9]. Hence, safety issues were highlighted which might be improved by minimizing the amount of the cyclodextrin. The sulfobutyl-β-cyclodextrin is more soluble and safer than hydroxypropyl-β-cyclodextrin, and, while many of these formulations showed improved pharmacodynamic profiles, there was often an increased duration of effect and stability problems.
Other propofol formulations use cosolvent mixtures with propofol solubilized in propylene glycol-water (1:1 v/v) or the prolinate ester of propofol and its water-soluble derivative dissolved in water at equimolar concentrations. However, studies with the prolinate ester in rats showed the formulation to have a longer induction time and longer duration of action [10].
A more recent water-based formulation of propofol is in a poly(N-vinyl-2-pyrrolidone) block polymer (Labopharm Inc., Quebec, Canada) in which propofol is dissolved inside micelles which are 30–60 nm in diameter [11, 12]. This solution is then lyophilized to form Propofol-PM (propofol polymeric micelle) that instantaneously reconstitutes to a clear solution upon addition of an aqueous medium. This formulation does not support the growth of microorganisms [11]. Micellar propofol has been examined in rats and shown to produce anesthesia. There were no differences in propofol kinetics compared with the “Diprivan” formulation [12]. There are no reported data on its use in higher species.
Propofol Analogs and Prodrugs
Other approaches have also been used to overcome some of the major disadvantages of propofol, specifically its hydrophobicity, cardiovascular and respiratory depression especially when given in association with opioids, and pain on injection. The physicochemical features of several propofol prodrugs have been described by Banaszczyk and colleagues [13]. The first attempt was a propofol sodium hemisuccinate prodrug, but this was unsuitable for commercialization as a stable aqueous solution. A phosphate ester formulation has been studied in mice, rats, rabbits, and pigs [13]. Metabolism by phosphatases yielded inorganic phosphate and propofol with a similar kinetic profile to the parent compound. Propofol concentrations greater than 1 ug.ml−1 were associated with sedation in rats and pigs. The median hypnotic dose of the propofol phosphate in mice was about ten times than seen when using propofol alone; and there was a similar increase in median lethal dose. Other highly water-soluble derivatives of propofol , formulated as cyclic amino acid esters, have been described [10]. The anesthetic properties of the most promising of these, the prolinate, have been described following intraperitoneal injection in rats and showed a faster loss of righting reflex than after “Diprivan.”
Fospropofol
Fospropofol (previously known as GPI-15715, Aquavan, Lusedra®) is the only clinically approved propofol analogue. It is a water-soluble phosphate ester of propofol (phosphono-O-methyl-2,6-diisopropylphenol). It is moderately rapidly and completely broken down in vivo to propofol , inorganic phosphate, and formaldehyde by alkaline phosphatases which are widely distributed in the body (Fig. 16.1). Based on the molecular weights of propofol and fospropofol, 1 mg of the latter should liberate 0.54 mg propofol . The formaldehyde is further broken down to formate which has not been shown to date to be associated with systemic toxicity.
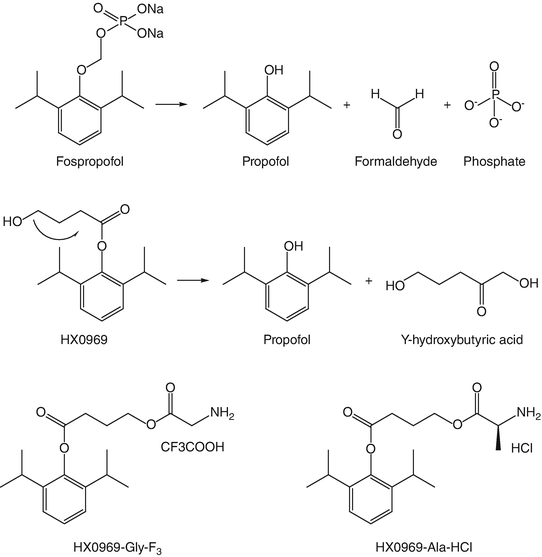
Fig. 16.1
Structures of fospropofol and HX0969w ; together with their metabolic breakdown pathways. Also shown are the structures of HX0969-Gly-F3 and HX0969-Ala-HCl
Administration of fospropofol is associated with a slow onset of effect (over 4–13 min depending on the dose used and much slower than the parent compound propofol —about 40 s). In man, the in vitro hydrolysis of fospropofol has a half-life of about 8 min. One of the issues to date is the absence of reliable pharmacokinetics (due to assay methodological problems). However preliminary data have suggested a short half-life (as might be expected from a drug undergoing plasma hydrolysis) and small apparent volume of distribution. The ED50 effective dose is about 6.5 mg kg−1, while the maximum recommended dose (12.5 mg kg−1) produces loss of consciousness in about 4 min.
The side effect profile of the drug is interesting—it has a lower incidence of hypotension, respiratory depression and apnea, and loss of airway patency than is seen with propofol at similar levels of sedation or anesthesia. This probably relates to the slower onset of effect of the drug. Although there have been no reports of pain on injection, a major side adverse effect is the onset of transient paresthesia and pruritus in the perineal region at the start of the infusion! Not surprisingly, these burning sensations were accompanied by a transient increase in heart rate [14]. A multicenter phase 3 trial has used lower doses of fospropofol (6.5 mg kg−1 instead of 5–14 mg kg−1) preceded by fentanyl 50 μg to produce moderate procedural sedation, followed by subsequent titrated doses of fospropofol [15]. Recovery was rapid and without any noticeable hangover effect. The drug has also been associated with a low incidence of postoperative nausea and vomiting. Comparison with propofol shows the new drug to have a lower incidence of adverse effects, with only minor respiratory depression. It has a slower onset of action than propofol , and the metabolic products cause no clinically significant drug-related sedative effects.
Paresthesia occurred in about 50 % of subjects, together with pruritus and perineal itching (although there was no apparent recall of the pain or discomfort). With this dose strategy, there was rapid recovery (with a median time to full alertness of 5 min and an Aldrete score >9 by 10 min) and improved patient outcome. In all studies reported to date, fospropofol has been associated with a low incidence of postoperative nausea and vomiting.
At the doses described above, 95 % of subjects have a blood propofol concentration <2 μg/ml. A single dose of fospropofol has a longer duration of effect compared with the same dose of propofol . It is not known to date whether prolonged use of fospropofol for sedation might lead to the propofol infusion syndrome.
Fospropofol has some apparent advantages over propofol and is associated with a lower risk of bacterial contamination as well as the absence of the infused lipid load that has been associated with organ toxicity during long-term infusions of Diprivan. Fospropofol was approved by the US FDA in December 2008 for use in monitored anesthesia care sedation in adult patients undergoing diagnostic or therapeutic procedures. But fospropofol can only be given by persons trained in the administration of general anesthesia and not involved in the conduct of procedure. Because of the slow-onset kinetics, the main utility of fospropofol will probably be for sedation in the intensive care unit, for procedural sedation outside of the operating room, and for sedation during monitored anesthesia care or regional anesthesia per se where its slow onset is less critical [16–18]. Present comparative studies have evaluated fospropofol against propofol and midazolam ; further studies are needed for the drug’s true profile needs to be better established.
Other New Chemical Entities Studied (To Date—2016) in Man
PF0713
PF0713 is a water-insoluble 2′6′dialkylphenol formulated as a 1 % lipid emulsion. It is a potent GABAA receptor agonist which, in the animal species so far tested, shows a hypnotic potency comparable to that of propofol , but with a slower onset of action and duration of effect. Structurally, it is a sterically hindered phenol (R,R 2′6′ di-sec-butylphenol) and is therefore similar in structure to propofol (Fig. 16.2). Indeed in its racemic form, it was one of the original compounds evaluated during the development of propofol .
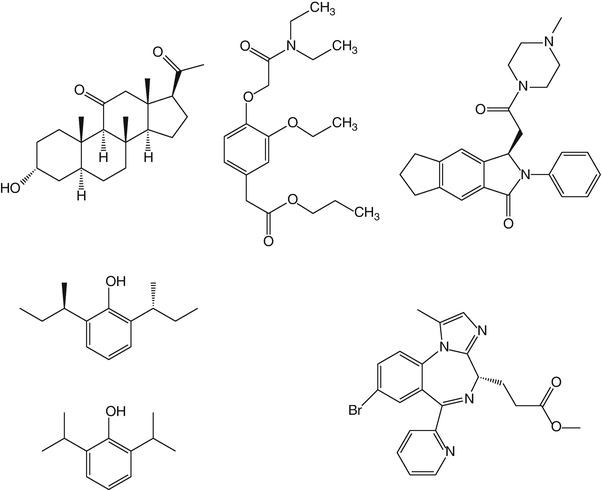
Fig. 16.2
Chemical structures of hypnotic drugs that have undergone evaluation in man. Top row—alfaxalone (Phaxan-CD), AZD 3043, and JM-1232 (also known as MR04A3). Second row—PF0713 (and structure of propofol for comparison) and remimazolam (CNS-7056X)
The actions of PF0713 have been examined at the molecular level using rat cerebral cortex binding assays, with the greatest effect found on the chloride channel (as has been shown for propofol ) and a lack of interaction with alpha2, NMDA, PCP, benzodiazepine, or opioid receptors [19]. In a comparison with the effects of propofol on the CA1 populations of pyramidal neurons, both drugs potentiated the effects of muscimol at the GABAA receptor, suggesting that the agent acts via the picrotoxin-binding site on the chloride binding channel.
When given to rats by the intravenous route, there is rapid onset of loss of the righting response, with a duration that was dose related over the range of 1.9–15.2 mg/kg. The drug appeared to be more potent than propofol in the rat, with the maximum tolerated dose being greater than that for propofol . However, after high doses (>7.0 mg/kg), PF0713 may be accompanied by prolonged recovery times [20].
Only one study has described the profile of PF0713 when administered to human subjects. At doses between 0.0156 and 2.0 mg/kg, there were no notable adverse events. At the higher doses of 1.0 mg/kg and 2.0 mg/kg, PF0713 produced rapid induction of general anesthesia without injection pain or agitation. When the depth and duration of anesthetic effect were assessed by the bispectral index and the Richmond Agitation and Sedation Score, the drug showed dose-related effects, with maintenance of blood pressure and heart rate. Preliminary kinetic data suggest that the drug has a high plasma clearance [21]. When given as a bolus dose, recovery was predictable. There are no data describing the drug’s profile when given by infusion; and further assessments of the agent have not been forthcoming.
JM-1232(−)
JM-1232(−) is one of a series of water-soluble sedative-hypnotics being evaluated by the Maruishi Pharmaceutical Company (Osaka, Japan) [22]. The lead compound [JM-1232(−)] is an isoindolin-1-one derivative which was shown to be active in mice (Fig. 16.2). It has a wide margin of safety—with a hypnotic ED50 of 3.1 mg/kg and LD50 of >120 mg/kg (giving a therapeutic index >35). In vitro binding data suggest the compound has a high affinity for the benzodiazepine receptor, but at a different binding site to midazolam . In vitro studies examining the effects of JM-1232(−) in a brain stem-spinal cord preparation of neonatal rats showed no effect on the C4 burst rate and amplitude at concentrations between 10 and 500 μM. However, at higher concentrations, there was depression of central respiratory activity which could be reversed by the addition of 100 μM flumazenil [23].
Recent studies have also examined the drug when given intrathecally to the rat, where it produces antinociception by an action thought to be on synaptic transmission at spinal nerves in the substantia gelatinosa. In vitro experiments suggest that JM-1232 enhances inhibitory transmission by prolonging the decay phase of the GABAergic sIPSCs. This effect might either be through benzodiazepine receptor activation or by increasing the spontaneous release of GABA and glycine from nerve terminals. This enhancement of the GABAergic effect could contribute to the antinociception seen in these animals [24].
JM-1232 has undergone limited clinical evaluation. When given as a 10 min infusion to male volunteers, it had a rapid onset and short duration of action [25]. Doses between 0.05 and 0.8 mg/kg caused sedation, with the higher doses producing a deeper and longer reduction in the bispectral index. There was no significant excitation associated with the onset of hypnosis (which is a common feature seen with neurosteroids and some other sedative-hypnotic agents). When given as a 1 or 10 min infusion at doses between 0.025 and 0.8 mg/kg, there were minimal effects on heart rate and blood pressure .
The drug produces dose-dependent sedation with rapid onset and recovery, although some volunteers showed upper airway obstruction at deeper levels of sedation. The population PK model indicates plasma clearance (for a 70 kg male) of 654 ml/min and apparent volume of distribution at steady state of 91.6 l per 70 kg. The plasma drug concentration associated with sedation was about 200 ng/mL−1 and the t 1/2 k e0 5.1 min [25]. Simulated context-sensitive half-times suggest a profile similar to propofol . Preliminary PK-PD data indicate an estimated EC50 effect-site concentration for anesthesia of 162 ng/mL and a t 1/2 k e0 of 4.2 min. The value for t 1/2 k e0 compares with 0.9 to 5.6 min for midazolam and 1.2–3.3 min for propofol . One unanswered question is whether the main metabolite of JM-1232 (so-called metabolite 3) has any hypnotic properties—especially at higher doses or during prolonged infusion.
Remimazolam (CNS 7056)
In animal studies, remimazolam is a benzodiazepine (Fig. 16.2) with a very predictable onset and offset of effect with a low risk of oversedation. The effects of the agonist are readily reversed by the benzodiazepine antagonist flumazenil. In vitro, remimazolam binds to benzodiazepine sites in the brain with a high affinity. Its carboxylic acid metabolite has a 300 times lower binding affinity. Neither the parent drug nor the main metabolite shows any binding affinity for other receptors, nor any selectivity for specific GABAA subtypes. In this respect, the pharmacological effects of remimazolam are similar to those of midazolam . Remimazolam causes dose-dependent inhibition of neuronal firing of the substantia nigra pars reticulata.
Remimazolam undergoes biotransformation by hydrolysis in human, rat, mouse, and minipig liver tissue, yielding CNS 7054 as the major metabolite. Metabolism also occurs in organs other than the liver (kidney, lung, brain), but there is no evidence of metabolism by plasma in human, minipig, or dog. The metabolite profile is therefore in keeping with remimazolam being a substrate for carboxylesterases rather than butyrylcholinesterase [26].
When given IV, 25 mg/kg midazolam and remimazolam both produced immediate loss of righting reflex in rats, but recovery of the righting reflex was faster with remimazolam [25 vs. 10 min, respectively]. The effect of remimazolam is inhibited by pretreatment with flumazenil [27]. In a kinetic and dynamic comparison with midazolam in the pigs [28], both drugs rapidly induced sedation, but recovery was faster after remimazolam. This slower recovery after midazolam can be related to the drug being eliminated more slowly (33 vs. 18 min half-life) and was more widely distributed with a larger apparent volume of distribution at steady state (1038 vs. 440 ml/kg), although the clearance rates of the two sedatives were similar (35.4 and 32.2 ml/kg/min, respectively).
Early human studies with remimazolam showed it to be well tolerated in doses between 0.05 and 0.35 mg/kg; at higher doses, episodes of hypoxia may be observed. However unlike other benzodiazepines, sedative doses are not accompanied by significant hypo- or hypertension. Plasma drug clearance is about 3× that of midazolam , and in studies to date, the kinetics are linear over a wide dose range. There was rapid onset of sedation (after 1 min with a peak at 4 min) and also rapid recovery (10 min compared with about 40 min for equipotent doses of midazolam ). At higher doses (>0.20 mg/kg), studies have reported episodes of blood oxygen desaturation [29]. A recent PK-PD assessment of remimazolam in doses from 0.01 to 0.3 mg/kg given by infusion over 1 min shows a population kinetic model with clearance of 66.7 l/h and apparent volume of distribution at steady state of 37.3 l, terminal half-life of 0.92 h, and mean residence time of 0.57 h [30]. The t 1/2 k e0 was 0.25 min−1 for remimazolam (compared with 0.05 min−1 for midazolam ). The IC50 for BIS was 0.26 μg/mL. Over the wide dose range studied to date, there is a linearity between dose and AUC to infinity.
On the basis of present knowledge, the CSHT for remimazolam appears to be short (15–16 min) for infusions lasting up to 3 h; this compares with values of 45–50 min for midazolam . However there are no presently published kinetics or dynamics of the agent in patients with liver and renal disease.
While this benzodiazepine was initially developed for use as a drug for procedural sedation, more recent studies have focused on use of the drug for both induction and maintenance of general anesthesia—especially within the fields of cardiovascular and cardiac anesthesia. When used in conjunction with analgesic agents (usually remifentanil), the drug has been compared with propofol . Remimazolam was infused at a dose of 1 mg/kg/h for maintenance after induction of anesthesia with an infusion rate of either 6 or 12 mg/kg/h. At these doses, all patients achieved satisfactory anesthesia for operative surgery. There were no major adverse effects, but the decrease in blood pressure following induction of anesthesia was about 50 % of that seen with the comparator propofol group. Recovery data are not presently available.
AZD3043
AZD3043 (previously known as THRX-918661 and TD4756) is a water-insoluble drug which is a congener of the hypnotic agent propanidid, but with about twice the potency of the latter. Its structure is that of an acetic acid propyl ester which is broken down through the action of plasma and tissue (mainly hepatic) esterases (Fig. 16.2). In vitro AZD 3043 undergoes rapid hydrolysis in whole blood of rat and guinea pig with half-lives of 0.4 and 0.1 min, respectively. There are no reported data on the activity of the main metabolites.
When given intravenously to rats, doses of 5–30 mg/kg caused dose-dependent loss of righting reflex and short-lived EEG suppression. After infusions ranging from 20 min to 5 h, the time to recovery of the righting reflex was about 3 min. In contrast, the recovery from propofol anesthesia ranged between 30 and 60 min. Faster recovery was also seen when AZD 3043 was compared with propofol in a pig model [31]. In the rat, an infusion of 2.5 mg/kg/min (in conjunction with remifentanil) was needed to maintain a surgical plane of anesthesia. After a 3 h continuous infusion in rats, kinetic studies showed a rapid loss of the parent compound within about 5 min. Comparable studies in the pig revealed that after a 3 h continuous infusion, clearance was about 3.4 l/kg/h and the elimination half-life was 0.4 h. Again, recovery was faster than after propofol when given at equipotent doses [31]. However one major concern about this agent is the low potency with doses for maintenance of anesthesia in the pig of the order of 1.5 mg/kg/min.
Clinical studies of the PK-PD relationships of remimazolam in man have been conducted in a single center series during which the pharmacology of the drug has been assessed based on bolus dosing (1–6 mg/kg) and 30 min infusions of the drug at doses ranging from 1 to 81 mg/kg/h [32–34]. The depth of sedation-hypnosis was measured using the BIS monitor. The aim of the studies was also to examine the sedation profile and safety of bolus doses and the combination of bolus and infusion to provide sedation (and anesthesia). The 30 min infusions of 1–81 mg/kg/h were associated with rapid onset of sedation or anesthesia at doses >12 mg/kg/h. All doses were accompanied by rapid and complete recovery although after the higher doses, recovery was associated with some involuntary movements and limb hypertonus.
Based on the blood concentration data from all 125 subjects, a kinetic model has been developed. This shows that in man AZD 3043 has a high clearance rate (with a mean value of 2.2 L/min) and a low apparent volume of drug distribution (which appears from the studies to date to be dose dependent ranging from about 15 L at low doses to 37 L at the highest doses). The main metabolites of AZD are the carboxylic acid metabolite (THRXZ-108893) and 1-propanol. Only a small fraction of the parent drug (0.02 %) is excreted unchanged. Breakdown occurs due to the action of both plasma and tissue (mainly hepatic) esterases with the metabolites being either inactive or showing only weak hypnotic properties. There is no evidence to date of any analgesic properties of the drug.
During the infusions of AZD 3043, drug effects on the processed EEG (BIS) were studied and the 50 % effective plasma concentration for hypnosis determined to be about 15 μg/mL (again, supporting the concept of the drug being of low potency). The blood-brain equilibration time was rapid (t 1/2 k eO about 1.1 min) but the slope of the concentration-effect relationship (gamma) was steep (1.7).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


