Neuromuscular Blocking Agents
François Donati
Key Points
Related Matter
Neuromuscular Blocking Agents
Fasciculations
Monitoring Nerve Block
For example, the incidence of awareness appears to be greater when neuromuscular blocking agents are used,3 and some authors recommend restricting the use of these drugs whenever possible, as patient movement might be an indicator of consciousness. However, anesthetics act at the spinal cord level to produce immobility; thus, movement in response to a noxious stimulus indicates inadequate analgesia and does not necessarily mean that the patient is conscious.4 Therefore, awareness does not occur because too much neuromuscular blocking agent has been given, but because too little anesthetic is administered. The controversy regarding neuromuscular blocking agents and awareness is complicated by the fact that neuromuscular blockade seems to affect the bispectral index (BIS), which is the most widely used measure of unconsciousness.5 Reductions in BIS have been reported in awake individuals receiving succinylcholine and in mildly sedated patients given mivacurium.
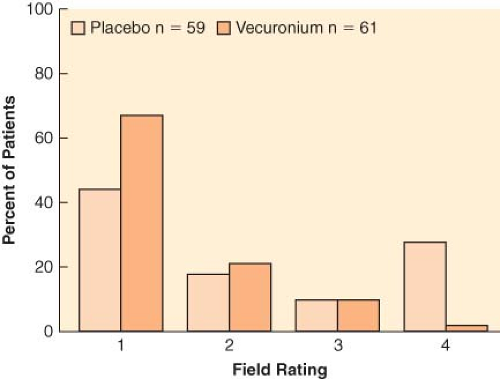 Figure 20.1. Surgeon’s assessment of muscle relaxation during lower abdominal surgery. Rating goes from 1 (excellent) to 4 (poor). The incidence of poor rating was greater in patients not given vecuronium (29%) compared with those who received the drug (2%). (Redrawn from: King M, Sujirattanawimol N, Danielson DR, et al. Requirements for muscle relaxants during radical retropubic prostatectomy. Anesthesiology. 2000;93:1392.) |
Complete paralysis is not required for the duration of all surgical procedures. However, neuromuscular blocking agents were found to make a difference in lower abdominal surgery, where surgical conditions were better in patients receiving vecuronium (Fig. 20-1).6 In addition to providing immobility and better surgical conditions, neuromuscular blocking agents improve intubating conditions. The doses of opioids required for acceptable intubating conditions in the absence of muscle paralysis produce significant hypotension (Fig. 20-2).7 Providing optimal intubating conditions is not a trivial objective. Poor intubating conditions may increase the incidence of laryngeal injury, as manifested by voice hoarseness and vocal cord damage (Fig. 20-3), and the best way to improve intubating conditions is to administer neuromuscular blocking agents.10
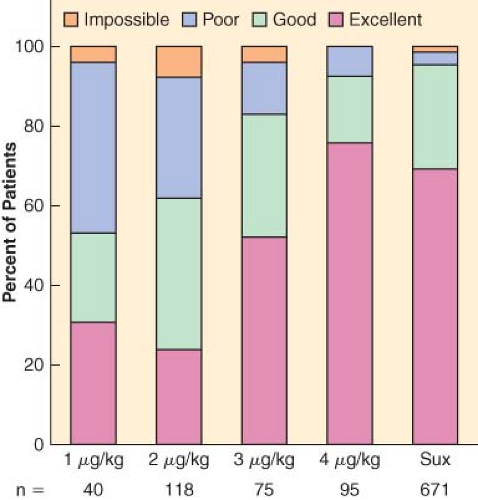 Figure 20.2. Neuromuscular blocking agents provide better intubating conditions than high doses of opioids, without hypotension. Hypnotic agent was propofol or thiopental. Intubating conditions are plotted against dose of remifentanil (in micrograms per kilogram). Results for succinylcholine (Sux), 1 mg/kg (with little opioid) are given for comparison. Hypotension was seen with remifentanil, 4 μg/kg. (Data obtained from references: McNeil IA, Culbert B, Russell I. Comparison of intubating conditions following propofol and succinylcholine with propofol and remifentanil 2 micrograms kg−1 or 4 micrograms kg−1. Br J Anaesth. 2000;85:623–625; Naguib M, Samarkandi AH, El-Din ME, et al. The dose of succinylcholine required for excellent endotracheal intubating conditions. Anesth Analg. 2006;102:151–155; Andrews JI, Kumar N, van den Brom RH, et al. A large simple randomized trial of rocuronium versus succinylcholine in rapid-sequence induction of anaesthesia along with propofol. Acta Anaesthesiol Scand. 1999;43:4–8; Durmus M, Ender G, Kadir BA, et al. Remifentanil with thiopental for tracheal intubation without muscle relaxants. Anesth Analg. 2003;96:1336; Klemola UM, Mennander S, Saarnivaara L. Tracheal intubation without the use of muscle relaxants: Remifentanil or alfentanil in combination with propofol. Acta Anaesthesiol Scand. 2000;44:465; and Donati F. The right dose of succinylcholine. Anesthesiology. 2003;99:1037) |
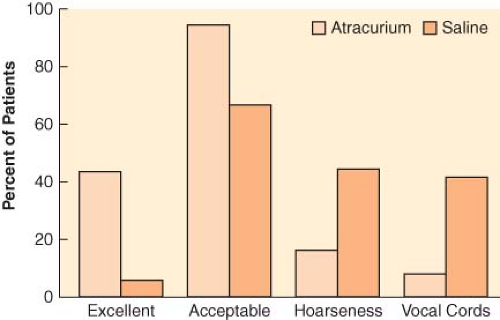 Figure 20.3. Neuromuscular blocking agents improve intubating conditions and reduce vocal cord sequelae. The graph depicts the incidence of excellent and acceptable (defined as good or excellent) intubating conditions after atracurium or saline. The percentage of patients who reported hoarseness and those with vocal cord lesions documented by stroboscopy is also shown. (Data from: Mencke T, Echternach M, Kleinschmidt S, et al. Laryngeal morbidity and quality of tracheal intubation: A randomized controlled trial. Anesthesiology. 2003;98:1049–1056.) |
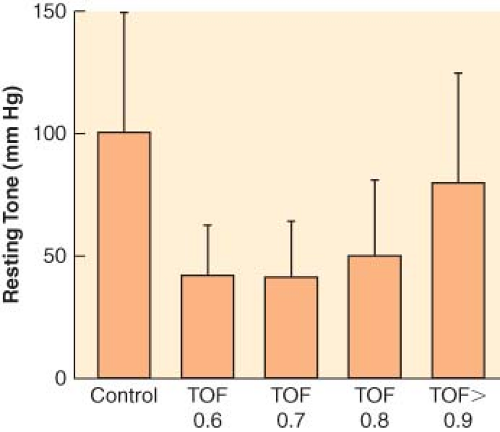 Figure 20.4. Upper esophageal resting tone in volunteers given vecuronium. Train-of-four (TOF) ratio was measured at the adductor pollicis muscle. Statistically significant decreases compared with control were found at all levels of paralysis until TOF ratio >0.9. (Redrawn from: Eriksson LI, Sundman E, Olsson R, et al. Functional assessment of the pharynx at rest and during swallowing in partially paralyzed humans: Simultaneous videomanometry and mechanomyography of awake human volunteers. Anesthesiology. 1997;87:1035.) |
Physiology and Pharmacology
Structure
The cell bodies of motor neurons supplying skeletal muscle lie in the spinal cord. They receive and integrate information from the central nervous system. This information is carried via an elongated structure, the axon, to distant parts of the body. Each nerve cell supplies many muscle cells (or fibers) a short distance after branching into nerve terminals. The terminal portion of the axon is a specialized structure, the synapse, designed for the production and release of acetylcholine. The synapse is separated from the endplate of the muscle fiber by a narrow gap, called the synaptic cleft, which is approximately 50 nm in width (0.05 μm) (Fig. 20-5).23,24,25 The nerve terminal is surrounded by a Schwann cell, and the synaptic cleft has a basement membrane and contains filaments that anchor the nerve terminal to the muscle.
Nerve Stimulation
Under resting conditions, the electrical potential of the inside of a nerve cell is negative with respect to the outside (typically −90 mV). If this potential is made less negative (depolarization), sodium channels open and allow sodium ions to enter the cell. This influx of positive ions makes the potential inside the membrane positive with respect to the outside. This potential change, in turn, causes depolarization of the next segment of membrane, causing more sodium channels to open, and an electrical impulse, or action potential, propagates. The duration of the action potential is brief (<1 ms) because of rapid inactivation of sodium channels and activation of potassium channels. An action potential also triggers the opening of calcium channels, allowing calcium ions to penetrate the cell. This entry of calcium facilitates release of the neurotransmitter at the nerve terminal.
The sodium channels in the axon may be activated in response to electrical depolarization provided by a nerve stimulator. A
peripheral nerve is made up of a large number of axons, each of which responds in an all-or-none fashion to the stimulus applied. Thus, in the absence of neuromuscular blocking agents, the relationship between the amplitude of the muscle contraction and current applied is sigmoid. At low currents, the depolarization is insufficient in all axons. As current increases, more and more axons are depolarized to threshold and the strength of the muscle contraction increases. When the stimulating current reaches a certain level, all axons are depolarized to threshold and propagate an action potential. Increasing current beyond this point does not increase the amplitude of muscle contraction: The stimulation is supramaximal (Fig. 20-6). Most commercially available stimulators deliver impulses lasting 0.1 to 0.2 ms.
peripheral nerve is made up of a large number of axons, each of which responds in an all-or-none fashion to the stimulus applied. Thus, in the absence of neuromuscular blocking agents, the relationship between the amplitude of the muscle contraction and current applied is sigmoid. At low currents, the depolarization is insufficient in all axons. As current increases, more and more axons are depolarized to threshold and the strength of the muscle contraction increases. When the stimulating current reaches a certain level, all axons are depolarized to threshold and propagate an action potential. Increasing current beyond this point does not increase the amplitude of muscle contraction: The stimulation is supramaximal (Fig. 20-6). Most commercially available stimulators deliver impulses lasting 0.1 to 0.2 ms.
Table 20-1. Selected Reports of Residual Paralysis 1979–2011 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Release of Acetylcholine
Acetylcholine is synthesized from choline and acetate and packaged into 45 nm vesicles. Each vesicle contains 5,000 to 10,000 acetylcholine molecules. Some of these vesicles cluster near the cell membrane opposite the crests of the junctional folds of the endplate, in areas called active zones (Fig. 20-5).23,24
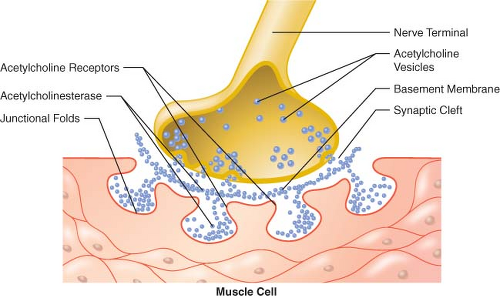 Figure 20.5. Schematic representation of the neuromuscular junction (not drawn to scale). |
It is now widely accepted that acetylcholine is released in packets, or quanta, and that a quantum represents the contents of one vesicle. In the absence of nerve stimulation, quanta are released spontaneously, at random, and this is seen as small depolarizations of the endplate (miniature endplate potential). When an action potential invades the nerve terminal, approximately 200 to 400 quanta are released simultaneously, unloading approximately 1 to 4 million acetylcholine molecules into the synaptic cleft.23,25 Calcium, which enters the nerve terminal through channels that open in response to depolarization, is required for vesicle fusion and release. Calcium channels are located near docking proteins, and this special geometric arrangement provides high intracellular concentrations of calcium to allow binding of specialized proteins on the vesicle membrane with docking proteins.23,24 Binding produces fusion of the membranes and release of acetylcholine ensues. When the calcium concentration is decreased, or if the action of calcium is antagonized by magnesium, the release process is inhibited and transmission failure may occur. Other proteins regulate storage and mobilization of acetylcholine vesicles. It appears that a small proportion of vesicles is immediately releasable, while a much larger reserve pool can be mobilized more slowly. Each impulse releases 0.2% to 0.5% of the 75,000 to 100,000 vesicles in the nerve terminal. With repetitive stimulation, the amount of acetylcholine released decreases rapidly because only a small fraction of the vesicles is in a position to be released immediately. To sustain release during high-frequency stimulation, vesicles must be mobilized from the reserve pool.
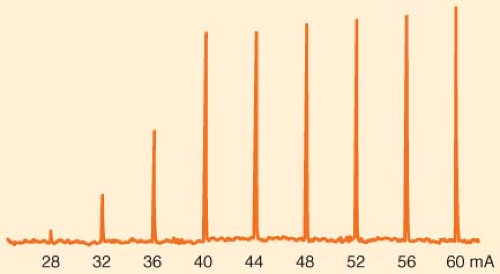 Figure 20.6. Example of increasing stimulating current in one patient. Current pulses, 0.2 ms duration, were delivered to the ulnar nerve at the wrist every 10 seconds. The force of contraction of the adductor pollicis muscle was measured and appears as spikes. No twitch was seen if the current was <28 mA. At current strengths of ≥40 mA, the current became supramaximal; increasing the current produced little change in force. |
Postsynaptic Events
The receptors at the endplate are of the nicotinic cholinergic type. They are members of the class of Cys-loop ligand-gated ion channels, to which glycine, 5-HT3, and GABA receptors belong.26 These receptors are made up of five glycoprotein subunits arranged in the form of a rosette and lying across the whole cell membrane (Fig. 20-7). To date, 17 subtypes of the nicotinic receptor have been identified, and they differ by the type and arrangement of the subunits.23 The nicotinic subtype present at the neuromuscular junction is made up of two identical subunits, designated α, and three others, called β, δ, and ε. There are two acetylcholine binding sites, each located on the outside part of the α subunit. When two acetylcholine molecules bind simultaneously to each binding site, an opening is created in the center of the rosette, allowing sodium ions to enter the cell and potassium ions to exit.23,24 The inward movement of sodium is predominant because it is attracted by the negative voltage of the inside of the cell. This movement of sodium depolarizes the endplate; that is, its inside becomes less negative. There is a high density of sodium channels in the folds of synaptic clefts and in the perijunctional area.24,25 These channels open when the membrane is depolarized beyond a critical point, allowing more sodium to enter the cell and producing further depolarization. This depolarization generates an action potential, which propagates by activation of sodium channels along the whole length of the muscle fiber. The muscle action potential has a duration of 5 to 15 ms and can be recorded as an electromyogram (EMG). It precedes the onset of contraction, or twitch, which lasts 100 to 200 ms. With high frequency (>10 Hz) stimulation, the muscle fiber does not have time
to relax before the next impulse, so contractions fuse and add up, and a tetanus is obtained.
to relax before the next impulse, so contractions fuse and add up, and a tetanus is obtained.
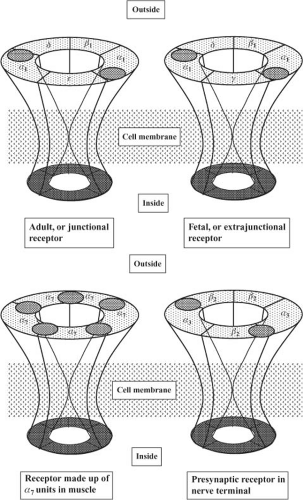 Figure 20-7. Four types of nicotinic receptors involved in neuromuscular transmission. All have five subunits. Postsynaptic adult receptors at the endplate have two α1, one β1, one δ, and one ε subunit. The immature, fetal or extrajunctional receptor has the same structure, except for a substitution of the ε by the γ subunits. It is distributed throughout the muscle membrane. Receptors made up of only α7 subunits are also found in muscle. Presynaptic receptors, found in the nerve terminal are thought to be of the α3β2 type. The acetylcholine binding sites are represented by a shaded oval area. |
Early in development, receptors are slightly different from those found at the adult endplate: They have a γ subunit instead of an ε subunit (Fig. 20-7), and they tend to be evenly distributed along the whole length of the muscle fiber.23,24,25 The receptors with a γ subunit, are called fetal receptors. When the endplate develops, receptors tend to cluster at the neuromuscular junction and the density is low (typically 10/μm2) in the extrajunctional areas.25 As maturation continues, the γ subunit is substituted by an ε subunit, which is characteristic of the adult type, junctional receptor. In humans, the switch occurs in the third trimester of pregnancy. During development, neuronal α7 receptors are also found in the muscle membrane. They are called neuronal because they are also found in the central nervous system, and they are made up of five identical α subunits, which differ slightly from the α subunits of the receptor at the neuromuscular junction, which are called α1. Maintenance of adult receptors at the endplate depends on the integrity of nerve supply. A few γ-type and α7 extrajunctional receptors still persist in adults and can proliferate in cases of denervation.23
The main action of nondepolarizing neuromuscular blocking drugs is to bind to at least one of the two α subunits of the postsynaptic receptor. This prevents access to the receptor by acetylcholine and does not produce opening of the receptor. Under normal circumstances, only a small fraction of available receptors must bind to acetylcholine to produce sufficient depolarization to trigger a muscle contraction. In other words, there is a wide
“margin of safety”.27 This redundancy implies that neuromuscular blocking drugs must be bound to a large number of receptors before any blockade is detectable. Animal studies suggest that 75% of receptors must be occupied before twitch height decreases in the presence of d-tubocurarine, and blockade is complete when 92% of receptors are occupied. The actual number depends on species and type of muscle, and humans might have a reduced margin of safety compared with other species.27 So it is futile to correlate receptor occupancy data obtained in cats with certain clinical tests in humans, such as handgrip and head lift, which involve different muscle groups. However, the general concept that a large proportion of receptors must be occupied before blockade becomes detectable, and that measurable blockade occurs over a narrow range of receptor occupancy, remains applicable to clinical practice. Because it must overcome the margin of safety, the initial dose of neuromuscular blocking agent is greater than maintenance doses.
“margin of safety”.27 This redundancy implies that neuromuscular blocking drugs must be bound to a large number of receptors before any blockade is detectable. Animal studies suggest that 75% of receptors must be occupied before twitch height decreases in the presence of d-tubocurarine, and blockade is complete when 92% of receptors are occupied. The actual number depends on species and type of muscle, and humans might have a reduced margin of safety compared with other species.27 So it is futile to correlate receptor occupancy data obtained in cats with certain clinical tests in humans, such as handgrip and head lift, which involve different muscle groups. However, the general concept that a large proportion of receptors must be occupied before blockade becomes detectable, and that measurable blockade occurs over a narrow range of receptor occupancy, remains applicable to clinical practice. Because it must overcome the margin of safety, the initial dose of neuromuscular blocking agent is greater than maintenance doses.
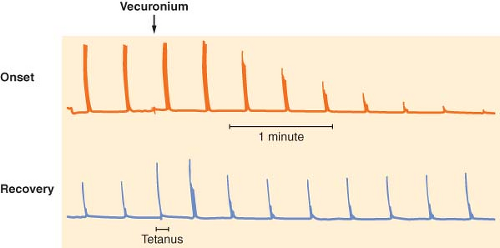 Figure 20.8. Characteristics of nondepolarizing blockade. Train-of-four (TOF) responses are equal before administration of vecuronium (arrow). For a given twitch depression, fade is less during onset (top trace) than recovery (bottom trace). A 50 Hz tetanus was applied during recovery, and the response is not sustained (tetanic fade). The TOF response immediately after the tetanus is enhanced, and progressively returns to its normal value within 1 to 2 minutes. |
Acetylcholine is hydrolyzed rapidly by the enzyme acetyl cholinesterase, which is present in the folds of the endplate. It is also attached to stalks attached to the basement membrane in the synaptic cleft. The presence of the enzyme in the synaptic cleft suggests that not all the acetylcholine released reaches the endplate; some is hydrolyzed en route.25,28
Presynaptic Events
The release of acetylcholine normally decreases during high frequency stimulation because the pool of readily releasable acetylcholine becomes depleted faster than it can be replenished. Under normal circumstances, the reduced amount released is well above what is required to produce muscle contraction because of the high margin of safety at the neuromuscular junction. In addition, a positive feedback system involving activation of presynaptic receptors helps in the mobilization of acetylcholine vesicles.23 There is some evidence that the presynaptic and postsynaptic receptors are of different subtypes, and that presynaptic receptors are most likely of the α3β2 subtype, that is, they are made up of only α and β subunits.29 This receptor contains α subunits that are slightly different from those found in postsynaptic receptors, thus the designation as α3, instead of the α1, and the β subunits (β2) are also slightly different from the β1 subunit found in postsynaptic receptors.
The putative role of the presynaptic receptors is to maintain the number of vesicles ready to be released. Nondepolarizing neuromuscular blocking drugs produce characteristic TOF and tetanic fade, probably by blocking presynaptic nicotinic receptors,23,29 thus preventing mobilization of acetylcholine vesicles and leading to reduced acetylcholine release during high frequency stimulation. Succinylcholine has virtually no effect on these presynaptic receptors, which would explain the lack of fade observed with this drug. Presynaptic receptors may not be the only elements contributing to the fade phenomenon: They do not explain why fade is more prominent during onset than during recovery (Fig. 20-8), and why after sugammadex, TOF ratio is restored faster than first twitch.30 Fade constitutes a key property of nondepolarizing neuromuscular blocking drugs and is useful for monitoring purposes.
Neuromuscular Blocking Agents
Pharmacologic Characteristics of Neuromuscular Blocking Agents
The effect of neuromuscular blocking drugs is measured as the depression of adductor muscle contraction (twitch) following electrical stimulation of the ulnar nerve. The value is compared with a control value, obtained before injection of the drug. Each drug has characteristic onset, potency, duration of action, and recovery index.
Potency of a drug is determined by constructing dose-response curves, which describe the relationship between twitch depression and dose (Fig. 20-9).31 Then, the effective dose 50, or ED50, which is the median dose corresponding to 50% twitch depression, is obtained. Because clinically useful relaxation is attained when twitch is abolished almost completely, the ED95, corresponding to 95% block, is more commonly used. For example, the ED95 for vecuronium is 0.05 mg/kg, which means that half the patients will achieve at least 95% block of single twitch (compared with the pre-vecuronium value) with that dose, and half the subjects will reach <95% block. Rocuronium has an ED95 of 0.3 mg/kg. Therefore, it has one-sixth the potency of vecuronium. In other words, compared with vecuronium, six times as much rocuronium has to be given to produce the same effect. The ED95
of known neuromuscular blocking agents vary over two orders of magnitude (Table 20-2).
of known neuromuscular blocking agents vary over two orders of magnitude (Table 20-2).
 Figure 20.9. Example of a dose-response relationship. The actual numbers are approximately those for rocuronium. The ED50 is the dose corresponding to 50% blockade and ED95 is the dose corresponding to 95% blockade. |
Onset time, or time to maximum blockade, can be shortened if the dose is increased. When two or more drugs are compared, it is meaningful to compare only equipotent doses and usually clinically relevant doses (2 × ED95) are considered.31
Duration of action is the time from injection of the neuromuscular blocking agent to return of 25% twitch height (compared with control). Duration increases with dose, so it is usual to make comparisons between 2 × ED95 doses, which are normal intubation doses. The 25% twitch height figure was chosen because rapid reversal can normally be achieved at that level. For nondepolarizing agents, the fourth twitch usually reappears when first twitch is close to 25%. Categories were proposed for neuromuscular blocking drugs according to their duration of action (Table 20-2).31 Agents with the same duration may have markedly different onsets.
Table 20-2. Potency, Onset Time, Duration, and Recovery Index of Selected Neuromuscular Blocking Agents | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Recovery index is the time interval between 25% and 75% twitch height. It provides information about the speed of recovery once return of twitch is manifest. Contrary to duration of action, it does not depend heavily on the dose given. The values for ED95, onset time, duration of action, and recovery index depend on which muscle is used to make measurements. For consistency, the adductor pollicis muscle has been retained as the gold standard, not because of its physiologic significance but because it is most commonly monitored and data on it are most abundant.
The pharmacologic characteristics of neuromuscular blocking agents are completed by an assessment of intubating conditions, which do not always parallel twitch height at the adductor pollicis muscle. Intubating conditions depend on paralysis of centrally located muscles, but also on the type and quantity of opioid and hypnotic drugs given for induction of anesthesia. To decrease variability between studies, criteria to grade intubating conditions as excellent, good, poor, or impossible in a scoring system were adopted by a group of experts who met in Copenhagen in 1994, and a revision of these standards was adopted at a later meeting in Stockholm in 2005.32
Depolarizing Drugs: Succinylcholine
Neuromuscular Effects
The effects of succinylcholine at the neuromuscular junction are not completely understood. The drug depolarizes postsynaptic and extrajunctional receptors. However, when the receptor is in contact with any agonist, including acetylcholine, for longer than a few milliseconds it ceases to respond to the agonist. Normally, this desensitization process does not occur with acetylcholine because of its rapid breakdown (<1 ms). However, succinylcholine remains at the endplate for much longer, so desensitization develops after a brief period of activation.33 Another possible mechanism is the inactivation of sodium channels in the junctional and perijunctional areas, which occurs when the membrane remains depolarized.33 This inactivation prevents the propagation of the action potential. Both desensitization of the receptor and inactivation of sodium channels might be present together.
Succinylcholine has yet another neuromuscular effect. In some muscles, like the masseter and to a lesser extent the adductor pollicis, a sustained increase in tension that may last for several minutes can be observed. The mechanism of action of this tension change is uncertain but is most likely mediated by acetylcholine receptors because it is blocked by large amounts of nondepolarizing drugs.35 The increase in masseteric tone, which is probably always present to some degree but is greater in some susceptible individuals, may lead to imperfect intubating conditions in a small proportion of patients. Masseter muscle spasm may be an exaggerated form of this response.
Characteristics of Depolarizing Blockade
After injection of succinylcholine, single-twitch height is decreased. However, the response to high frequency stimulation is sustained: Minimal TOF and tetanic fade is observed. The block is antagonized by nondepolarizing agents so that the ED95 is doubled if a small “defasciculating” dose of nondepolarizing drug is given prior to the intubating dose of succinylcholine, thus requiring an increased dose of succinylcholine.36 Succinylcholine blockade is potentiated by inhibitors of acetyl cholinesterase, such as neostigmine and edrophonium. After administration of 7 to 10 mg/kg, or 30 to 60 minutes of exposure to succinylcholine, TOF and tetanic fade become apparent. Neostigmine or edrophonium can antagonize this block, which has been termed nondepolarizing, dual, or phase II block. The onset of phase II block coincides with tachyphylaxis, as more succinylcholine is required for the same effect.
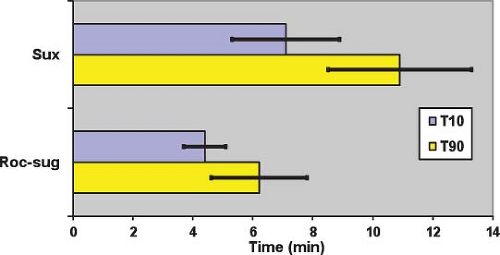 Figure 20.10. Duration of action of neuromuscular blockade with succinylcholine 1 mg/kg (Sux), and rocuronium 1.2 mg/kg followed 3 minutes later by sugammadex 16 mg/kg (Roc-sug). T10 and T90: Duration to 10% and 90% first twitch recovery, respectively. Bars indicate standard deviation. Data from: Lee C, Jahr JS, Candiotti KA, et al. Reversal of profound neuromuscular block by sugammadex administered three minutes after rocuronium. Anesthesiology. 2009;110:1020–1025. |
Pharmacology of Succinylcholine
Succinylcholine is rapidly hydrolyzed by plasma cholinesterase (also called pseudocholinesterase), with an elimination half-life of <1 minute in patients.37 Because of the rapid disappearance of succinylcholine from plasma, the maximum effect is reached quickly. Sub-paralyzing doses (up to 0.3 to 0.5 mg/kg) reach their maximal effect within approximately 1.5 to 2 minutes at the adductor pollicis muscle,36 and within 1 minute at more central muscles, such as the masseter, the diaphragm, and the laryngeal muscles. With larger doses (1 to 2 mg/kg), abolition of twitch response can be reached even more rapidly (<1 minute).
The mean dose producing 95% blockade (ED95) at the adductor pollicis muscle is 0.30 to 0.50 mg/kg.36 These values are doubled if d-tubocurarine, 0.05 mg/kg, is given as a defasciculating agent.36 The time until full recovery at the adductor pollicis is dose-dependent and reaches 10 to 12 minutes after a dose of 1 mg/kg (Fig. 20-10).38 The diaphragm starts contracting and spontaneous breathing resumes after approximately 5 minutes.39
Side Effects
Cardiovascular
Sinus bradycardia with nodal or ventricular escape beats (or both) may occur, especially in children, and asystole has been described after a second dose of succinylcholine in both pediatric and adult patients. These effects can be attenuated with atropine or glycopyrrolate.40 The mechanisms for the cardiovascular side effects of succinylcholine are not known because succinylcholine appears to have little effect on autonomic cholinergic receptors.33 Succinylcholine increases catecholamine release, and tachycardia is seen frequently.
Anaphylaxis
Succinylcholine has been incriminated as the trigger of allergic reactions more often than any other drug used in anesthesia. Successive studies conducted in France indicate that the number of reported events is decreasing, corresponding to the gradual
replacement of succinylcholine by nondepolarizing drugs.41 The incidence of anaphylactic reactions to succinylcholine is difficult to establish, but is probably of the order of 1:5,000 to 1:10,000.
replacement of succinylcholine by nondepolarizing drugs.41 The incidence of anaphylactic reactions to succinylcholine is difficult to establish, but is probably of the order of 1:5,000 to 1:10,000.
Fasciculations
The prevalence of fasciculations is high (60% to 90%) after the rapid injection of succinylcholine, especially in muscular adults. This is a benign side effect of the drug, but many clinicians prefer to prevent fasciculations with a small dose of a nondepolarizing neuromuscular blocking drug is given 3 to 5 minutes before succinylcholine is effective.34 When the drug was available, d-tubocurarine 0.05 mg/kg, or 3 mg, was used for this purpose. Rocuronium is an acceptable alternative, as long as appropriate doses (0.03 to 0.04 mg/kg, or 10% of the ED95) are given.42 A dose of 0.06 mg/kg leads to an unacceptably high incidence of symptoms of neuromuscular weakness, such as blurred vision, heavy eyelids, voice changes, difficulty swallowing, or even dyspnea, in the awake patient.34,42,43 A meta-analysis shows that these side effects have been observed frequently,34 but most of these data come from studies where large defasciculating doses of neuromuscular blocking agents, exceeding 10% of the ED95, were given.42 Atracurium, 0.02 mg/kg, is also effective. Pancuronium, vecuronium, cisatracurium, and mivacurium are not as effective as defasciculants. After these nondepolarizing drugs, the dose of succinylcholine must be increased from 1 mg/kg to 1.5 or preferably 2 mg/kg because of the antagonism between depolarizing and nondepolarizing drugs.36 Other drugs, such as diazepam, lidocaine, fentanyl, calcium, vitamin C, magnesium, dantrolene, or a small dose of succinylcholine have all been used to prevent fasciculations. The results are no better than with nondepolarizing drugs and they may have undesirable effects of their own.
Muscle Pains
Generalized aches and pains, similar to the myalgia that follows violent exercise, are common 24 to 48 hours after succinylcholine administration. Their incidence is variable (1.5% to 89% of patients receiving succinylcholine) and are more common in young, ambulatory patients.44 The intensity of muscle pains is not always correlated with the intensity of fasciculations, but the methods that have been shown effective to prevent fasciculations usually prevent muscle pains. For example, a precurarization dose of a nondepolarizing neuromuscular blocking agent is effective. Lidocaine (1 to 1.5 mg/kg), especially in conjunction with precurarization, has also been shown to be of value.44 Calcium, vitamin C, benzodiazepines, magnesium, and dantrolene have been tried with inconclusive results.44
Intragastric Pressure
Succinylcholine increases intragastric pressure, and this effect is blocked by precurarization. However, succinylcholine causes even greater increases in lower esophageal sphincter pressure. Thus, succinylcholine does not appear to increase the risk of aspiration of gastric contents unless the lower esophageal sphincter is incompetent.
Intraocular Pressure
Intraocular pressure increases by 5 to 15 mm Hg after injection of succinylcholine, and this increase is still present after detachment of extraocular muscle, suggesting an intraocular etiology. Precurarization with a nondepolarizing blocker has little or no effect on this increase. This information has led to the widespread recommendation to avoid succinylcholine in open-eye injuries. However, it must be appreciated that inadequate anesthesia, elevated systemic blood pressure, and insufficient neuromuscular blockade during laryngoscopy and tracheal intubation might increase intraocular pressure more than succinylcholine. In addition, there is little evidence that the use of succinylcholine has led to blindness or extrusion of eye content.45
Intracranial Pressure
Succinylcholine may increase intracranial pressure, and this response is probably diminished by precurarization.46 Again, laryngoscopy and tracheal intubation with inadequate anesthesia or muscle relaxation are likely to increase intracranial pressure even more than succinylcholine.
Hyperkalemia
Serum potassium increases by approximately 0.5 mEq/L a few minutes after injection of succinylcholine. This increase is not prevented completely by precurarization. In fact, only large doses of nondepolarizing blockers reliably abolish this effect.47 Subjects with preexisting hyperkalemia, such as patients in renal failure, do not have a greater increase in potassium levels, but the absolute level might reach the toxic range. Succinylcholine is safe in normokalemic renal failure patients.48 Severe hyperkalemia, occasionally leading to cardiac arrest, has been described in patients after major denervation injuries, spinal cord transection, peripheral denervation, stroke, trauma, extensive burns, and prolonged immobility with disease, and may be related to potassium loss via a proliferation of extrajunctional receptors.47 Hyperkalemia has been reported with myotonia and muscle dystrophies, and cardiac arrests have been reported in children before the diagnosis of the disease was made.47 Severe hyperkalemia after succinylcholine resulting in cardiac arrest has also been observed in acidotic hypovolemic patients.
Abnormal Plasma Cholinesterase
Plasma cholinesterase activity can be reduced by a number of endogenous and exogenous causes, such as pregnancy, liver disease, uremia, malnutrition, burns, plasmapheresis, and oral contraceptives. These conditions usually lead to a slight, clinically unimportant increase in the duration of action of succinylcholine. Plasma cholinesterase activity is reduced by the anticholinesterases neostigmine and pyridostigmine, but not edrophonium, so that the duration of succinylcholine given after neostigmine or pyridostigmine, but not after edrophonium, is increased.49
The cholinesterase gene is located on chromosome 3 at q26,50 and over 20 mutations in the coding region of the plasma cholinergic gene have been identified. Many of these mutations are relatively frequent, like the K (13%) variants, but do not lead to clinically significant increases in the duration of blockade after succinylcholine. Other alleles, such as the A, F, or silent variant, are less frequent, and are associated with significant prolongation of succinylcholine blockade only if the patient is homozygous for the allele, or if the allele is associated with another abnormal allele. Only approximately 1:2,000 individuals have prolonged paralysis (2 to 6 hours) after usual doses of succinylcholine (1 to 1.5 mg/kg). In heterozygous patients with an abnormal allele combined with the usual allele (1:30 cases), the duration of action is only slightly prolonged compared with normal individuals. Traditional methods for identifying plasma cholinesterase phenotype involve measurement of enzyme activity with a substrate and inhibition with dibucaine, fluoride, and chloride. These tests are only capable of identifying some enzyme variants. The complete amino acid sequence of plasma cholinesterase has now been
determined using molecular genetics techniques.50 Whole blood or fresh-frozen plasma can accelerate succinylcholine metabolism in patients with low or absent plasma cholinesterase, but the best course of action is probably mechanical ventilation of the lungs and providing adequate sedation to the patient until full recovery of neuromuscular function can be demonstrated.
determined using molecular genetics techniques.50 Whole blood or fresh-frozen plasma can accelerate succinylcholine metabolism in patients with low or absent plasma cholinesterase, but the best course of action is probably mechanical ventilation of the lungs and providing adequate sedation to the patient until full recovery of neuromuscular function can be demonstrated.
Clinical Uses
The main indication for succinylcholine is to facilitate tracheal intubation. In adults, a dose of 1.0 mg/kg yields 75% to 80% excellent intubation conditions within 1 to 1.5 minutes after an induction sequence that includes a hypnotic (propofol or thiopental) and a moderate opioid dose.8,51 The dose must be increased to 1.5 to 2.0 mg/kg if a precurarizing dose of nondepolarizing blocker has been used.36 Intubating conditions without precurarization are only marginally improved by increasing the dose to 2 mg/kg.8
Succinylcholine is especially indicated for “rapid sequence induction,” when a patient presents with a full stomach and the possibility of aspiration of gastric contents. In this situation, manual ventilation of the lungs is avoided, if possible, to reduce the probability of aspiration because of excessive intragastric pressure caused by gas forced via the face mask. Thus, the ideal neuromuscular blocking agent has both a fast onset, to reduce the time between induction and intubation of the airway, and a rapid recovery, to allow return of normal breathing before the patient becomes hypoxic. The duration of action of succinylcholine, given at a dose of 1 mg/kg, is short enough so that the majority of properly preoxygenated patients resume respiratory efforts (5 to 6 minutes) before hypoxia can be detected.38,39 It has been argued that this is valid only in relatively healthy subjects and not in all cases. As a result, a lower dose has been suggested. However, a dose of 0.5 to 0.6 mg/kg results in substantially fewer patients with excellent intubating conditions, and the decrease in duration is modest.8 For maintenance of relaxation, typical infusion rates are approximately 50 to 100 μg/kg/min. However, the availability of short and intermediate nondepolarizing drugs makes succinylcholine infusions obsolete.
Children are slightly more resistant to succinylcholine than adults,52 and doses of 1 to 2 mg/kg are required to facilitate intubation. In infants, 2 to 3 mg/kg may be required. Precurarization is not necessary in patients younger than 10 years because fasciculations are uncommon in this age group. Bradycardia is common in children unless atropine or glycopyrrolate is given.40 Succinylcholine, at a dose of 4 mg/kg, is the only effective intramuscular neuromuscular blocking agent in children with difficult intravenous access and provides adequate intubating conditions in about 4 minutes. However, this route of administration should not be the method of choice.53
In obese individuals, the dose of succinylcholine, in milligrams per kilogram of actual body weight, is the same as in leaner patients. Calculating the dose per kilogram ideal body weight (IBW) might lead to underdosing and inadequate intubating conditions.54 The volume of distribution, expressed per kilogram of actual body weight, of succinylcholine is probably decreased in obese individuals, but this is compensated by an increase in plasma cholinesterase activity.
Nondepolarizing Drugs
Nondepolarizing neuromuscular blocking drugs bind to the postsynaptic receptor in a competitive fashion, by binding to one of the α subunits of the receptor (Fig. 20-7).24
Characteristics of Nondepolarizing Blockade
Finally, nondepolarizing blockade can be antagonized with anticholinesterase agents like neostigmine. It is also antagonized by depolarizing agents such as succinylcholine provided that the nondepolarizing blockade is intense and that the succinylcholine dose is too small to produce a block of its own.
Pharmacokinetics
As is the case for other drugs used in anesthesia, the elimination half-life of neuromuscular blocking agents does not always correlate with duration of action because termination of action sometimes depends on redistribution instead of elimination. However, knowledge of the kinetics of the drug helps us understand the behavior of the drug in special situations (prolonged administration, disease of the organs of elimination, and so on).
Several mechanisms can explain the various categories of durations of action listed in Table 20-3:
All long-duration drugs have a long (1 to 2 hours) elimination half-life and depend on liver and/or kidney function for termination of action. Return of twitch height occurs during the elimination phase of the drug.
Intermediate-duration drugs either have an intermediate elimination half-life or long elimination half-lives but depend on redistribution rather than elimination for termination of effect. Atracurium and cisatracurium have intermediate elimination half-lives (20 to 25 minutes) and recovery of twitch height occurs during the elimination phase. Vecuronium and rocuronium have longer terminal half-lives (approximately 90 minutes), but their relatively short duration of action is due to extensive redistribution of the drug, so that recovery of twitch height occurs during redistribution, rather than during the elimination phase. Thus, durations of action of vecuronium, rocuronium, atracurium, and cisatracurium are similar.
Short-duration drugs have short elimination half-lives. The active isomers of mivacurium have elimination half-lives of a few minutes.
Ultrashort-duration drugs have a very short elimination half-life. Succinylcholine has an elimination half-life of less than 1 minute.
The volume of distribution of all these agents is approximately equal to extracellular fluid (ECF) volume (0.2 to 0.4 L/kg; Table 20-3). In infants, in whom the ECF volume as a proportion of body weight is increased, the volume of distribution of neuromuscular blocking drugs parallels ECF volume closely.
Table 20-3. Typical Pharmacokinetic Data for Neuromuscular Blocking Agents in Adults, Except Where Stated | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Onset and Duration of Action
Both onset time and duration of action depend on the dose given, but even at equipotent doses, different drugs may have different onset times and duration of action, because other factors play a role.
Onset time is determined by the time required for drug concentrations at the site of action to reach a critical level, usually that corresponding to 100% block. Onset time (2 to 7 minutes) is longer than time to peak plasma concentrations (<1 minute). This delay reflects the time required for drug transfer between plasma and neuromuscular junction and is represented quantitatively by a rate constant (keo). This rate constant corresponds to half-times of 5 to 10 minutes for most nondepolarizing drugs and is determined by all the factors that modify access of the drug to, and its removal from, the neuromuscular junction. These include cardiac output, distance of the muscle from the heart, and muscle blood flow. Thus, neuromuscular blockade usually occurs sooner at well-perfused centrally located muscles (diaphragm or vocal cords) than at peripheral locations (hand or foot). Also, if metabolism or redistribution is very rapid, for example, in the case of succinylcholine, the onset time is accelerated. Finally, potent drugs have a slower onset of action than less potent agents (Fig. 20-11).57 This is because a large proportion of receptors must be occupied before blockade can be observed. Blockade of these receptors will occur faster, and onset will be more rapid, if more drug molecules
are available, that is, if potency is low. Table 20-2 shows that onset tends to be slower if a drug is potent, that is, if ED95 is small.
are available, that is, if potency is low. Table 20-2 shows that onset tends to be slower if a drug is potent, that is, if ED95 is small.
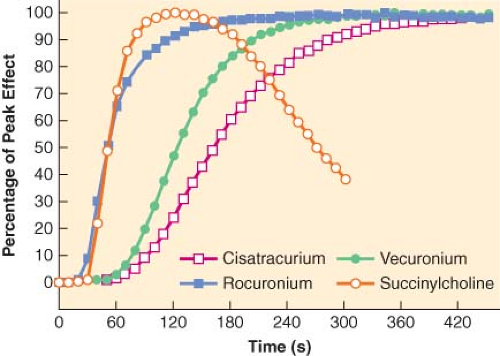 Figure 20.11. Neuromuscular blockade as a function of time for four neuromuscular blocking agents. Onset is faster for the less potent succinylcholine and rocuronium than for the more potent vecuronium and cisatracurium. (From: Kopman AF, Klewicka MM, Kopman DJ, et al. Molar potency is predictive of the speed of onset of neuromuscular block for agents of intermediate, short, and ultrashort duration. Anesthesiology. 1999;90:425, with permission.) |
Duration of action is determined by the time required for drug concentrations at the site of action to decrease below a certain level, usually corresponding to 25% first twitch blockade. For a drug with rapidly decreasing plasma concentrations, duration of action tends to be short; for a drug that is eliminated slowly from plasma, duration of action will be long.
Individual Nondepolarizing Agents
Since 1942, nearly 50 nondepolarizing neuromuscular blocking agents have been introduced into clinical anesthesia. This section covers only those drugs currently available in North America and Europe, plus a few others of historical interest. The first agent to undergo clinical investigation was intocostrin, or d-tubocurarine, the purified and standardized product of curare obtained from the plant Chondodendron tomentosum.1 d-tubocurarine has been completely replaced by more modern synthetic analogues.
d-tubocurarine
The dose of d-tubocurarine required to produce 95% twitch block at the adductor pollicis muscle, or ED95, is 0.5 mg/kg. At that dose, the duration of action is typical of a long-duration agent (Table 20-2). The molecule undergoes minimal metabolism and is excreted in the kidney and in the bile. Like most other neuromuscular blocking drugs, it is not extensively (30% to 50%) protein bound. Hypotension frequently accompanies the administration of d-tubocurarine even at doses <ED95. The mechanism involved is mainly histamine release, and skin flushing is frequently observed. Autonomic ganglionic blockade may also play a minor role.
Pharmacokinetics
d-tubocurarine was the first neuromuscular blocking agent whose concentration could be measured in plasma, and the pharmacokinetic studies conducted with this drug have led to the development of concepts that are helpful in understanding the behavior of all nondepolarizing agents in patients of different age groups and with disease of organs of elimination. The volume of distribution of the drug, expressed in mL/kg body weight, is greater in infants than in older children, young adults, and the elderly, but ED50 of d-tubocurarine, expressed in mg/kg body weight, does not vary markedly with age, from infancy to old age. These findings suggest that the d-tubocurarine concentration required for a given degree of blockade is less in infants than in older patients (Table 20-3).58 This phenomenon has also been observed with other neuromuscular blocking agents. The decreased glomerular filtration rate in the very young and the very old results in an increased elimination half-life and prolonged duration of action.58 The onset of action is more rapid in the young as a result of a more rapid circulation time.
Burns
Patients with massive burns demonstrate resistance to d-tubocurarine and other nondepolarizing drugs that depends on the size of the burn and the time since injury.59 Higher concentrations of the free drug are required to produce a given degree of twitch depression compared with nonthermally injured patients. Compared with normal subjects, the number of acetylcholine receptors is increased in muscles close to the site of burn injury, but also, to a lesser extent, in more distant muscles.60
Clinical Use
The long duration and cardiovascular effects of d-tubocurarine have restricted its use. Drugs of intermediate duration with virtually no cardiovascular effects have almost eliminated the use of d-tubocurarine. When available, d-tubocurarine has been mainly confined to be used as a “precurarization” (3 mg/70 kg) before succinylcholine to reduce fasciculations and muscle pains. Rocuronium has largely replaced d-tubocurarine for this indication.
Atracurium
Atracurium is a bisquaternary ammonium benzylisoquinoline compound of intermediate duration of action. It is degraded via two metabolic pathways. One of these pathways is the Hofmann reaction, a nonenzymatic degradation with a rate that increases as temperature and/or pH increases. The second pathway is nonspecific ester hydrolysis. The enzymes involved in this metabolic pathway are a group of tissue esterases, which are distinct from plasma or acetyl cholinesterases.61 The same group of enzymes is involved in the degradation of esmolol and remifentanil. It has been estimated that two-thirds of atracurium is degraded by ester hydrolysis and one-third by Hofmann reaction. Subjects with abnormal plasma cholinesterase have a normal response to atracurium.
The end products of the degradation of atracurium are laudanosine and acrylate fragments. Laudanosine has been reported as causing seizures in animals, but at doses largely exceeding the clinical range. No deleterious effect of laudanosine has been demonstrated conclusively in humans.61 Laudanosine is excreted by the kidney. Acrylates have been shown to inhibit human cell proliferation in vitro.62 However, the concentrations and exposure times required to obtain this effect are much greater than what is obtained normally in clinical practice.
Pharmacology
Atracurium is an intermediate-duration drug, with a terminal half-life of approximately 20 minutes. Termination of effect occurs during the elimination phase of the drug. Duration of action does not depend on age, renal function, or hepatic function. The ED95 of atracurium is 0.2 to 0.25 mg/kg. The onset of action is 3 to 5 minutes at 2 × ED95 (0.5 mg/kg). Onset of atracurium can be shortened if the dose is increased, but it is not recommended to exceed 0.5 mg/kg because of hypotension and histamine release. The duration of action is also dose-related. The time to 25% first twitch recovery after 0.5 mg/kg is approximately 30 to 40 minutes.
Cardiovascular Effects
Like d-tubocurarine, atracurium releases histamine in a dose-related manner. If large doses (≥0.5 mg/kg) are administered, hypotension, tachycardia, skin flushing, and possibly bronchospasm may occur. These manifestations, which are observed in virtually every subject given a large enough dose, should not be confused with an anaphylactic reaction, which is a rare event seen in a few susceptible individuals, but could be triggered by a small dose. Anaphylactic reactions to atracurium have been described, but they do not appear to be more frequent than after other neuromuscular blocking drugs.41
Special Situations
Dosage requirements are similar in the elderly, younger adults, and children, presumably reflecting the organ independence of atracurium’s elimination. Similarly, no dosage adjustment is required in
individuals with renal or hepatic failure. As with other nondepolarizing agents, the dose must be increased in burn patients, partly because of increased protein binding and partly because of upregulation of receptors, causing resistance at the endplate. In the obese patient, the dose of atracurium, as for all neuromuscular blocking agents, should be calculated on the basis of lean body mass.
individuals with renal or hepatic failure. As with other nondepolarizing agents, the dose must be increased in burn patients, partly because of increased protein binding and partly because of upregulation of receptors, causing resistance at the endplate. In the obese patient, the dose of atracurium, as for all neuromuscular blocking agents, should be calculated on the basis of lean body mass.
Clinical Uses
Atracurium has fallen into disfavor, probably because of its cardiovascular effects, in the United States, but remains popular in certain parts of Europe.63 Intubating doses are 0.5 mg/kg, and laryngoscopy should be attempted only after 2 to 3 minutes. Cardiovascular manifestations of histamine release are often seen at that dose, and perfect intubating conditions are seen in only half the patients (Fig. 20-3).10 For intubation, there has been a tendency to replace atracurium by agents with a shorter onset time and more cardiovascular stability, such as rocuronium. However, atracurium is convenient and versatile for maintenance of relaxation, either as a continuous infusion (5 to 10 μg/kg/min) or as intermittent injections (0.05 to 0.1 mg/kg every 10 to 15 minutes).
Cisatracurium
In an attempt to increase the gap between the neuromuscular blocking dose and the histamine-releasing dose, a potent isomer of atracurium, cisatracurium, was identified. Like atracurium, its cardiovascular effects are manifest only at doses exceeding 0.4 mg/kg, but its ED95 (0.05 mg/kg) is much lower. As a result, manifestations of histamine release are not seen in practice. The metabolism of cisatracurium is similar to that of atracurium, with Hofmann and ester hydrolysis both playing a role.61
Pharmacology
Because cisatracurium is a potent drug, its onset time is longer than that of atracurium and longer still than that of rocuronium. For example, equipotent doses of cisatracurium (0.046 mg/kg) and rocuronium (0.31 mg/kg) had onset times of 4.5 minutes and 1.7 minutes, respectively.57 The elimination half-life (22 to 25 minutes) is similar to that of atracurium,64 so the duration of action for 2 × ED95 doses (0.1 mg/kg) is 30 to 45 minutes. However, in an attempt to accelerate onset, the recommended intubating dose is increased to 0.15 mg/kg. This dose is well below the threshold for histamine release, but the duration of action is prolonged to 45 to 60 minutes. Because the doses required to obtain paralysis are considerably less for cisatracurium than for atracurium, less laudanosine and less acrylate byproducts are produced.61,62 Thus, the concerns raised by the potential toxic effects of these metabolites are virtually eliminated.
Special Situations
Like atracurium, there is no need to adjust dosage in the elderly, children, or infants, when compared with young adults. The experience in burn patients is limited, but the same principles that are valid for atracurium are expected to apply. In obese individuals, the dose of cisatracurium should be calculated on the basis of IBW.65
Side Effects
In contrast to atracurium, cisatracurium is devoid of histamine-releasing properties even at high doses (8 × ED95). It is also devoid of cardiovascular effects. However, anaphylactic reactions have been described.41
Clinical Use
Cisatracurium may be used to facilitate tracheal intubation at doses equivalent to 3 to 4 times the ED95 (0.15 to 0.2 mg/kg) when manual ventilation is possible after induction of anesthesia and when the duration of the procedure is expected to exceed 1 hour. Duration is shorter with lower doses, but onset time is prolonged and intubating conditions are less ideal. Neuromuscular blockade is easily maintained at a stable level by continuous intravenous infusion of cisatracurium (1 to 2 μg/kg/min) at a constant rate and does not change with time, suggesting the lack of a significant cumulative drug effect and lack of dependence on renal and/or hepatic clearance mechanisms.66 The rate of recovery is independent of the dose of cisatracurium and the duration of the administration.
Since cisatracurium does not depend on end-organ function for its elimination, the drug appears suitable for administration in the intensive care unit (ICU). The infusion rates to keep patients paralyzed are greater than in the operating room (typically 5 μg/kg/min), with wide interindividual variability.67 It is likely that prolonged exposure of the receptors to a neuromuscular blocking agent causes some upregulation, with a corresponding requirement for a higher dose.59 Interestingly, patients with acute respiratory distress syndrome requiring respiratory support in the ICU randomized to cisatracurium had better survival than patients receiving placebo.68
Doxacurium
Doxacurium is a potent, long-acting benzylisoquinoline compound that is not degraded by Hofmann elimination or ester hydrolysis. Its elimination half-life is 1 to 2 hours and it depends on the kidney and the liver for its disposition. Thus, duration of action is prolonged in the elderly and in subjects with impaired renal or hepatic function. The ED95 for doxacurium is 25 μg/kg (Table 20-2).69 Its cardiovascular stability is useful, but it has a limited place in clinical practice because of its very slow onset and long duration of action. It is unsuitable for facilitating tracheal intubation or for providing skeletal muscle relaxation during brief surgical procedures. When used in the ICU, recovery after stopping the infusion is prolonged when compared with cisatracurium.
Gantacurium and Other Chlorofumarates
Gantacurium was the first compound belonging to the class of asymmetric mixed-onium chlorofumarates to be investigated. It is a nondepolarizing drug and belongs to the class of asymmetric mixed-onium chlorofumarates. Its main degradation pathway involves cysteine in the plasma and is independent of plasma cholinesterase. The ED95 in humans is approximately 0.19 mg/kg.70 Cardiovascular effects are observed at doses exceeding 3 × ED95, and are most probably related to histamine release. At doses anticipated to be required for tracheal intubation (0.4 to 0.6 mg/kg), onset at the adductor pollicis muscle is 1.5 minutes and duration to 25% T1 recovery is 8 to 10 minutes, comparable with that of succinylcholine.
Other compounds of the same class have also been synthesized, with the idea of developing a compound with more cardiovascular stability, longer duration of action, and the ability to be reversed by cysteine.70 The most promising of these compounds is now CW002, for which the concept has been tested in dogs.71
Mivacurium
Mivacurium is a benzylisoquinoline derivative with a short duration of action that is hydrolyzed by plasma cholinesterase, like
succinylcholine.72 Contrary to succinylcholine, however, mivacurium produces nondepolarizing blockade. The drug is presented as a mixture of three isomers. Two of the isomers, the cis–trans and trans–trans, have short half-lives, but the cis–cis isomer has a much longer half-life (Table 20-3). The pharmacology of mivacurium is governed largely by the behavior of the trans–trans and cis–trans isomers, because the cis–cis isomer accounts for only 6% of the mixture and is less potent than the other two isomers. Mivacurium is no longer available in North America, but is used in many European countries.63
succinylcholine.72 Contrary to succinylcholine, however, mivacurium produces nondepolarizing blockade. The drug is presented as a mixture of three isomers. Two of the isomers, the cis–trans and trans–trans, have short half-lives, but the cis–cis isomer has a much longer half-life (Table 20-3). The pharmacology of mivacurium is governed largely by the behavior of the trans–trans and cis–trans isomers, because the cis–cis isomer accounts for only 6% of the mixture and is less potent than the other two isomers. Mivacurium is no longer available in North America, but is used in many European countries.63
Pharmacology
The ED95 of mivacurium has been estimated in the range of 0.08 to 0.15 mg/kg (Table 20-2). At 0.2 or 0.25 mg/kg doses, intubating conditions are not as good as with succinylcholine. Onset time is surprisingly long for a drug whose active isomers have a terminal half-life of <2 minutes, with disappearance of the twitch taking as long as 2.5 to 4 minutes at 2 to 3 × ED95.72 This long onset time is probably the result of the high potency of mivacurium.57 Recovery to 25% does not depend heavily on dose, being in the range of 15 to 25 minutes for doses of 0.15 to 0.25 mg/kg. The infusion rate to maintain blockade constant does not vary markedly with time, and recovery is as rapid after many hours of infusion than after a bolus dose.72,73
Side Effects
Like atracurium, mivacurium releases histamine in a dose-related fashion. Hypotension, tachycardia, and cutaneous signs, such as erythema and flushing, are seen frequently when doses exceed 0.2 mg/kg.
Pediatric Population
In infants and children the ED95 is approximately the same as in adults, but onset of block and recovery are more rapid. Cardiovascular effects are not as important as in adults, so doses up to 0.3 mg/kg have been used. The infusion rate required to maintain blockade is greater in children than in adults, and less in the elderly than in younger adults.
Burns
In burn patients, upregulation of the receptors, and to a lesser extent increased protein binding, causes a resistance to all nondepolarizing neuromuscular blocking agents. However, for mivacurium, the situation is different because plasma cholinesterase activity is decreased in burn patients. The net effect is either a normal or even an enhanced effect of usual doses.74
Reversal
Plasma Cholinesterase
Mivacurium is metabolized by plasma cholinesterase somewhat more slowly than succinylcholine. The conditions associated with a decreased plasma cholinesterase activity known to affect succinylcholine metabolism also alter mivacurium duration of action.
Clinical Use
Mivacurium is well suited to surgical procedures requiring brief muscle relaxation, particularly those in which rapid recovery is required, such as ambulatory and laparoscopic surgery. However, it is not recommended for rapid-sequence induction. Cardiovascular effects may be avoided by administering the drug slowly or by splitting the dose into two injections, 30 seconds apart. Maintenance of relaxation is accomplished more easily by constant infusion (5 to 7 μg/kg/min in young and middle-aged adults) than by intermittent bolus injection. This infusion rate has to be increased in children and reduced in the elderly.
Pancuronium
Pancuronium belongs to a series of bisquaternary aminosteroid compounds. It is metabolized to a 3-OH compound, which has one-half the neuromuscular blocking activity of the parent compound. The ED95 of pancuronium is 0.07 mg/kg. The duration of action is long, being 1.5 to 2 hours after a 0.15 mg/kg dose. Clearance is decreased in renal and hepatic failure, demonstrating that excretion depends on both organs. The onset of action is more rapid in infants and children than in adults, and recovery is slower in the elderly.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


