NEUROLOGIC EMERGENCIES
MARC H. GORELICK, MD, MSCE AND MATTHEW P. GRAY, MD, MS
GOALS OF EMERGENCY CARE
Signs and symptoms of neurologic dysfunction in children are produced by either primary nervous system disorders or are secondary to systemic disease. The differential diagnosis of many such neurologic findings can be found in the second section of this book (see related chapters below). This chapter focuses on the management of conditions primarily involving the various parts of the nervous system, including the brain, spinal cord, and peripheral nerves. Most neurologic conditions present with a recognizable pattern of clinical manifestations. Goals of care in the emergency department should focus on prompt recognition of these patterns, systematic diagnostic evaluations, and appropriate subspecialist consultation.
KEY POINTS
 A detailed history and neurologic examination are imperative to identifying the etiology of most neurologic conditions.
A detailed history and neurologic examination are imperative to identifying the etiology of most neurologic conditions.
 Anatomic localization of a neurologic injury is usually possible after evaluation of the distribution and character of the deficit.
Anatomic localization of a neurologic injury is usually possible after evaluation of the distribution and character of the deficit.
 Neuroimaging is a useful adjunct in the diagnosis of many neurologic diseases and MRI is often the study of choice.
Neuroimaging is a useful adjunct in the diagnosis of many neurologic diseases and MRI is often the study of choice.
RELATED CHAPTERS
Signs and Symptoms
• Dizziness and Vertigo: Chapter 19
• Eye: Visual Disturbances: Chapter 25
Medical, Surgical, and Trauma Emergencies
SEIZURES (SEE ALSO CHAPTER 67 SEIZURES)
Goals of Treatment
Most seizures in children are brief, lasting less than 5 minutes. Status epilepticus was classically defined as seizures that are continuous for 30 minutes or longer or repetitive seizures between which the patient does not regain consciousness. Many authorities now consider that seizures lasting for longer than 5 minutes or multiple seizures with no return to baseline in between constitute early status epilepticus. Initial management of the child with a seizure is directed at preventing neurologic damage through supportive care and seizure termination with timely administration of antiepileptic drugs, as well as identifying any treatable underlying cause of the seizure.
CLINICAL PEARLS AND PITFALLS
• Status epilepticus should be treated aggressively to minimize neurologic damage.
• Routine laboratory studies and acute neuroimaging are unnecessary for the majority of children with first-time seizures.
Current Evidence
Epidemiologic studies indicate that 3% to 6% of children will have at least one seizure in the first 16 years of life; most of these are simple febrile seizures, which generally occur in children 6 months to 6 years old. A seizure is a transient, involuntary alteration of consciousness, behavior, motor activity, sensation, and/or autonomic function caused by an excessive rate and hypersynchrony of discharges from a group of cerebral neurons. The term convulsion is often used to describe a seizure with prominent motor manifestations. Epilepsy, or seizure disorder, is a condition of susceptibility to recurrent seizures.
The basic pathophysiologic abnormality common to all seizures and convulsions is the hypersynchrony of neuronal discharges. Many precipitating factors, including metabolic, anatomic, and infectious abnormalities (see Chapter 67 Seizures), may produce seizures. Seizures that result from an identified precipitant are called symptomatic, or provoked, seizures, whereas those with no precipitating factor are called idiopathic or cryptogenic. Febrile seizures (seizures occurring in association with a febrile illness, without evidence of intracranial infection or other identified cause) are a particular type of provoked seizure seen in children between the ages of 6 months and 6 years. The exact cause of febrile seizures remains elusive. Elevated body temperature lowers the seizure threshold, and the immature brain appears to have a particular susceptibility to seizures in response to fever. Individual predisposition plays an important role.
During a seizure, cerebral blood flow, oxygen and glucose consumption, and carbon dioxide and lactic acid production increase dramatically. If the patient remains well ventilated, the increase in cerebral blood flow is sufficient to meet the increased metabolic requirements of the brain. Brief seizures rarely produce lasting deleterious effects on the brain; however, prolonged and serial seizures, especially status epilepticus, may be associated with permanent neuronal injury.
Clinical Considerations
Clinical Recognition
When the physician is examining a child with an acute paroxysmal event, the first step is to distinguish seizures from other nonepileptic phenomena. If the event is indeed a seizure, it may be classified according to type. Finally, a specific causative factor should be sought. The extent of the emergency evaluation is determined by the clinical scenario; some of the diagnostic assessment may be deferred. Of course, when a child is actively seizing, the first priority is to provide necessary resuscitation measures and control the seizures (see Chapter 67 Seizures and the following sections).
Paroxysmal events other than seizures that involve changes in consciousness or motor activity are common during childhood and may mimic epilepsy (Tables 67.1 and 105.1). Breath-holding spells occur in children 6 months to 4 years of age. Breath-holding spells take two forms: cyanotic and pallid. In the cyanotic form, the infant begins crying vigorously, often in response to an inciting event, then holds his or her breath and becomes cyanotic. After approximately 30 to 60 seconds, the child becomes rigid. As the spell ends, the child becomes limp and may have a transient loss of consciousness and twitching or jerking of the extremities, but the child quickly returns to full alertness. A pallid breath-holding spell may follow a seemingly insignificant trauma. The child may start to cry, but then turns pale and collapses. There is a brief period of apnea and limpness, followed by rapid recovery. In both types of breath-holding spells, the typical history and lack of postictal drowsiness help determine the diagnosis. Breath-holding spells may be recurrent but disappear spontaneously before school age.
Syncope is a brief, sudden loss of consciousness and muscle tone. There are numerous causes of syncope, many of which can be detected on the basis of historical information, physical examination, and simple diagnostic tests (see Chapter 71 Syncope). A syncopal episode can usually be distinguished from a seizure on the basis of the description. The child is typically upright before the event and often senses a feeling of light-headedness or nausea. The child then becomes pale and slumps to the ground. The loss of consciousness is brief, and recovery is rapid. This is in contrast to seizures, which usually have a postictal period with sleepiness. On awakening, the child is noted to have signs of increased vagal tone, such as pallor, clammy skin, dilated pupils, and relative bradycardia. Patients with narcolepsy also experience sudden alterations in alertness, with sleep occurring suddenly and uncontrollably during the daytime. In about half of the patients, narcolepsy is associated with cataplexy, an abrupt loss of muscle tone brought on by a sudden emotional outburst. Narcolepsy is far less common than syncope; both occur more often in adolescents than in younger children.
TABLE 105.1
NONEPILEPTIC EVENTS THAT MAY MIMIC SEIZURES

Single episodes of staring, involuntary movements, or eye deviation have been found to occur commonly in the first months of life, although they rarely lead to the parent seeking medical attention. In some children, however, these episodes occur frequently. Children with benign shuddering attacks have episodes of staring and rapid tremors involving primarily the arms and head, sometimes associated with tonic posturing. The episode lasts only a few seconds, and afterward, the child resumes normal activity. Acute dystonia, usually seen as a side effect of certain medications, can mimic a tonic seizure. The child having a dystonic reaction, however, does not lose consciousness and has no postictal drowsiness.
Several paroxysmal events are associated with sleep. Night terrors (see Chapter 134 Behavioral and Psychiatric Emergencies) usually begin in the preschool years. The sleeping child wakes suddenly, is confused and disoriented, and appears frightened, often screaming and showing signs of increased autonomic activity (tachycardia, tachypnea, sweating, dilated pupils). Such episodes typically last only a few minutes, and the child does not usually recall the event. Benign myoclonus is characterized by self-limited episodes of sudden jerking of the extremities, usually upon falling asleep. There is no alteration of consciousness. In sleep paralysis, there is a transient inability to move during the transition between sleeping and waking, also with no change in the level of consciousness.
Pseudoseizures are occasionally seen, often in patients with an underlying seizure disorder or in patients who have a relative with epilepsy. Some features suggestive of pseudoseizures are suggestibility; lack of coordination of movements; moaning or talking during the seizure; lack of incontinence, autonomic changes, or postictal drowsiness; response to painful stimuli; and poor response to treatment with anticonvulsant agents.
TABLE 105.2
SEIZURE TYPES
The most important diagnostic test in distinguishing nonepileptic events from seizures is a careful history, including a detailed description of the event from the person who witnessed it. In atypical or unclear cases, referral for electroencephalogram (EEG) or video EEG monitoring may help in establishing the diagnosis.
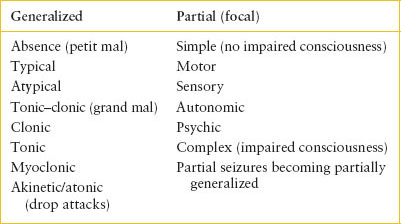
Clinically, seizures may be divided into partial (also termed focal) and generalized seizures (Table 105.2), and partial seizures are further classified as complex or simple. Complex partial seizures imply impaired consciousness. Generalized tonic–clonic seizures (previously called grand mal seizures) are the type most often seen in acute pediatric care. The onset of generalized tonic–clonic seizures is usually abrupt, although 20% to 30% of children may experience a sensory or motor aura. If sitting or standing, the child falls to the ground. The face becomes pale, the pupils dilate, the eyes deviate upward or to one side, and the muscles contract. As the increased tone of the thoracic and abdominal muscles forces air through the glottis, a grunt or cry may be heard. Incontinence of urine or stool is common. After this brief tonic phase (10 to 30 seconds), clonic movements occur. The child is unresponsive during the seizure and remains so postictally for a variable period. After the seizure, there may be weakness or paralysis of one or more areas of the body (Todd paralysis). In atonic, or akinetic, seizures (drop attacks), there is abrupt loss of muscle tone and consciousness. Myoclonic seizures are characterized by a sudden dropping of the head and flexion of the arms (jackknifing); however, extensor posturing may also occur. The episodes occur quickly and frequently, as often as several hundred times daily.
Absence (petit mal) seizures are generalized seizures, marked by sudden and brief loss of awareness, usually lasting 5 to 30 seconds. With typical absence seizures, there is no loss of posture or tone and no postictal confusion. There may be a minor motor component such as eyelid blinking.
The child with simple partial (focal) seizures has unimpaired consciousness. Motor signs are most common in children, although sensory, autonomic, and psychic manifestations are possible. The motor activity usually involves the hands or face and spreads in a fixed pattern determined by the anatomic origin of the nerve fibers that innervate the various muscle groups. Focal seizures may become secondarily generalized, in which case there will be alteration of consciousness. Complex partial seizures, also called psychomotor or temporal lobe seizures, exhibit a diverse set of clinical features, including alterations of perception, thought, and sensation. In children, they are usually marked by repetitive and complex movements with impaired consciousness and postictal drowsiness.
An important distinction is whether the seizure is associated with fever. Simple febrile seizures are those that are single, brief (lasting less than 15 minutes), and generalized. Approximately 20% of febrile seizures are complex, meaning they are focal, prolonged (last for more than 15 minutes), or multiple episodes within 24 hours.
Triage and Initial Assessment
For an actively seizing child, initiate immediate resuscitative measures and consider administration of antiepileptic agents, as discussed below. After seizures have stopped, the first steps in the evaluation are a thorough history and a physical examination, the results of which are helpful in determining the direction of the search for a specific cause (see Table 67.1 and Fig. 67.1). Important historical items to elicit include fever, trauma, underlying illnesses, current medications, and possible toxic ingestions. A complete neurologic assessment to evaluate for signs of increased intracranial pressure (ICP), focal deficits, or signs of meningeal irritation is also essential.
Diagnostic Testing
In children older than 12 months with a typical simple febrile seizure and no signs of meningitis, generally no further evaluation of the seizure is required. However, lumbar puncture (LP) is indicated if meningitis is suspected on the basis of physical findings. An LP should be considered in children younger than 12 months, in whom signs of meningitis may be subtle, such as irritability and poor feeding; when the febrile seizure is complex; or if there has been pretreatment with antibiotics. In addition, LP should be considered for children with prolonged fever before the seizure, and for febrile children who do not return to neurologic baseline quickly. Other laboratory tests discussed in the next paragraph have been found to have little yield in the child with a typical febrile seizure and are unnecessary. Appropriate diagnostic tests to determine the source of the fever are determined by other features such as the intensity of fever, immunization status, and the child’s age.
For the child who presents with a first-time, nonfebrile seizure, laboratory or radiologic evaluation to search for a specific treatable cause of the seizure may be indicated. There is little utility in extensive, routine workups; rather, ancillary test selection should be guided by the results of the history and physical examination. In young infants, children with prolonged seizures, and those with a suggestive history or physical examination, determination of serum glucose, sodium, and calcium levels are indicated. Other ancillary tests that may be indicated, depending on the clinical picture, include serum magnesium, hepatic transaminases, ammonia, serum or urine toxicology tests, electrocardiogram (ECG), and neuroimaging of the brain. LP is rarely emergently necessary in the afebrile child without meningeal signs or altered mental status, although it should be considered in neonates even without fever.
In children with a known seizure disorder, subtherapeutic anticonvulsant levels are the most common reason for breakthrough seizures. The name and dosage of anticonvulsant medications used should be elicited, as well as the time of the last dose given, any missed doses, the last change in dosage, and recent levels, if known. Intercurrent illness may also play a role because the metabolism of some medications is affected by systemic illness. Such children should have blood drawn for measurement of anticonvulsant levels. Although many drugs have a standard therapeutic range (Table 105.3), individual patients may require levels outside that range for adequate seizure control; conversely, dose-dependent toxic effects may be observed in some children even at typically therapeutic levels.
TABLE 105.3
COMMONLY USED ANTICONVULSANT AGENTS
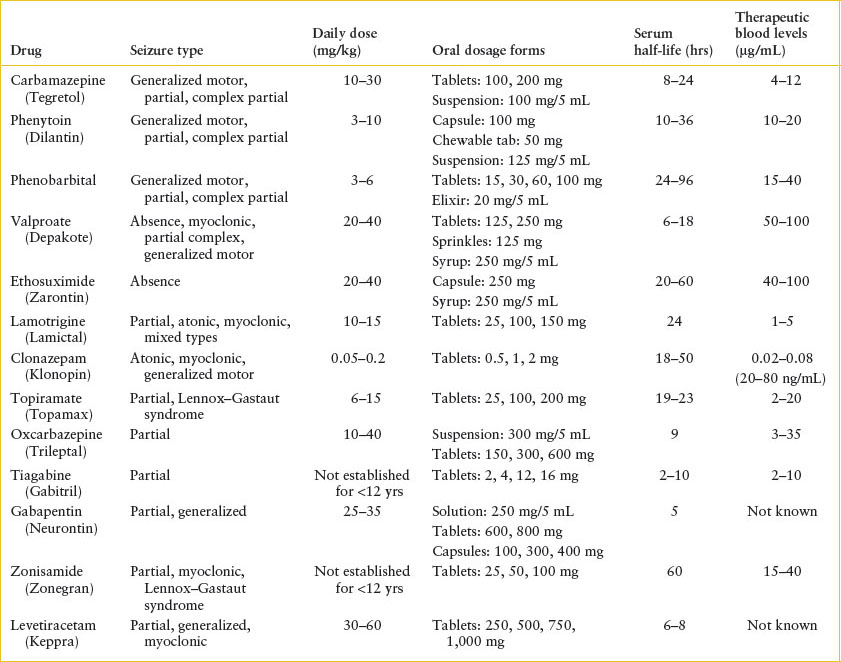
Computed tomography (CT) (or magnetic resonance imaging [MRI], if available) is indicated in the emergency evaluation of prolonged or focal seizures, when focal deficits are present, when there is a history of trauma, when the child has a ventriculoperitoneal shunt, or when there are associated signs of increased ICP. For other children with a normal neurologic examination, MRI may be useful in identifying structural anomalies and determining prognosis, but such studies may be deferred to a follow-up visit. Cranial imaging is not indicated in the evaluation of simple febrile seizures. EEG is also helpful in the evaluation of children with nonfebrile seizures. EEG is rarely beneficial in acute management, but children with nonfebrile seizures should be referred for outpatient testing.
Management
The administration of supplemental oxygen and maintenance of an adequate airway are vital parts of the initial management of the unconscious, actively convulsing child (see Chapter 67 Seizures). Trismus often occurs in generalized seizures but is transient. If the teeth are tightly clenched, even the placement of the oral airway should be deferred until it can be inserted during a phase of relaxation to avoid trauma. Seizure-associated hypoventilation and apnea are common with prolonged seizures, and may be a side effect of anticonvulsant medications; thus providers caring for such children should be prepared to offer assisted ventilation. For patients with adequate ventilatory effort but unable to fully maintain their airway, consideration should be given to airway adjuncts and airway positioning. Intravenous (IV) access should be established promptly; however, because of the potential for increased ICP, fluid therapy should be used judiciously until a more thorough evaluation is performed. The child with active convulsions should be protected from trauma.
It is unusual for the child with a brief seizure to arrive in the ED actively convulsing because, by definition, such seizures last for less than 15 minutes. Therefore, the actively convulsing child is usually already in a prolonged or serial seizure state, and pharmacologic intervention to terminate the seizure is required. Establish IV access, and draw blood for diagnostic studies. If hypoglycemia is documented by rapid glucose assay or if rapid determination is unavailable, give IV glucose in a dose of 2 to 4 mL per kg of 25% dextrose in water, or 5 to 10 mL per kg of 10% dextrose (use only the latter in infants). If hyponatremia is suspected based on a history of frequent vomiting or diarrhea or dilution of infant formula, emergent point-of-care testing for sodium should be performed. Seizures caused by hypoglycemia or hyponatremia are unlikely to be treated successfully with anticonvulsant medications without addressing the underlying cause. In neonates or in children with suspected isoniazid toxicity, IV pyridoxine 100 mg may be administered.
In most situations, benzodiazepines are the first drug of choice for acute seizures because of their rapidity of action. Overall effectiveness is approximately 70% in children. Lorazepam (Ativan) is the historically preferred agent, although recent evidence has not demonstrated superiority over Versed (midazolam) or Valium (diazepam). Given in a dose of 0.1 mg per kg IV (usual maximum 4 mg per dose), it has an onset of action of 2 to 5 minutes, and the duration of anticonvulsant effect is 12 to 24 hours. Half the dose may be repeated after 5 minutes. An alternative is diazepam (Valium), 0.2 to 0.4 mg per kg IV (usual maximum 10 mg per dose), which has a similarly rapid onset of action. If IV or intraosseous access cannot be established, diazepam may be administered rectally in a dose of 0.5 mg per kg (maximum 20 mg per dose), instilling the IV formulation with a slip-tip syringe (remove needle) or by using a specific rectal gel preparation. Intramuscular (IM) midazolam (Versed) has also been shown to be effective in a dose of 0.2 mg per kg (maximum 7 mg per dose). IV midazolam may also be given; the intranasal and buccal routes have also been described and appear to have promising initial results.
All the benzodiazepines can cause sedation and respiratory depression, which may persist for hours. Equipment for establishing an airway and supporting respiration must be available, especially if repeated doses are used. Hypotension is uncommon but may be a problem with multiple doses or when barbiturates are administered concomitantly.
If the seizures have not been controlled within 10 minutes with benzodiazepines, fosphenytoin (or phenytoin, if fosphenytoin is unavailable) should be given. Fosphenytoin is a prodrug of phenytoin, which is rapidly metabolized to the active form. It offers several advantages over phenytoin, including more rapid administration and fewer local and systemic side effects. Fosphenytoin may also be given IM, unlike phenytoin. The dose of the two drugs is identical; fosphenytoin doses are expressed as phenytoin equivalents (PE). The loading dose of fosphenytoin is 15 to 20 mg PE per kg IV, at a rate of 3 mg PE/kg/min to a maximum of 150 mg per minute. In the absence of any clinical effect, an additional 10 mg per kg IV may be administered. Cardiac monitoring is required because rapid IV infusion may lead to hypotension, QT prolongation, and cardiac dysrhythmias. (If phenytoin is used instead, the maximum rate of administration is 2 mg/kg/min in children and 50 mg per minute in an adult.) In patients known to be taking phenytoin chronically, a smaller dose of 5 to 10 mg per kg should be used initially unless the serum level is known to be very low. Each 1 mg per kg of phenytoin administered raises the serum level by approximately 1 μg per mL, although phenytoin kinetics are unpredictable. Phenytoin is highly lipid soluble and reaches therapeutic levels in the brain within 10 to 20 minutes, with duration of action of 12 to 24 hours. Unlike other anticonvulsant medications, phenytoin does not cause sedation or respiratory depression.
Phenobarbital is the next agent often added if phenytoin is not effective or contraindicated (e.g., allergy, known therapeutic level). The loading dose of phenobarbital is 20 mg per kg, sometimes given in two divided doses. The total drug dose is given over 5 to 10 minutes IV (maximum 30 mg per minute in an adult), or IM in the absence of IV access. Onset of action is usually within 15 to 20 minutes and lasts more than 24 hours. Phenobarbital, like other barbiturates, may cause significant sedation, respiratory depression, and hypotension.
IV valproate (VPA) and IV levetiracetam (Keppra) may serve as alternatives to phenobarbital or phenytoin in the treatment of SE. Valproic acid may be particularly useful for patients with a known seizure disorder, who are currently using VPA, when low serum concentrations are suspected. Effective loading dose in one study was 10 mg per kg IV when subtherapeutic levels were suspected and 25 mg per kg IV when the patient was not being treated with VPA. In this study, no adverse effects related to hypotension or heart rate were observed. The loading dose of levetiracetam is 20 to 60 mg per kg.
Patients with SE that lasts for more than 30 to 60 minutes present a special problem. Further management should be done, when possible, in conjunction with a neurologist and with EEG monitoring. Continuous infusion of benzodiazepines, barbiturate coma, or general anesthesia may be used. Previously, paraldehyde and lidocaine hydrochloride have been used to treat SE. However, these medications have not demonstrated any advantage over the conventional therapies already discussed and have more serious side effect profiles.
With prolonged seizures, the duration of postictal drowsiness and confusion may also be protracted. However, the child who fails to arouse within 15 to 30 minutes after cessation of seizures should be evaluated carefully to rule out nonconvulsive SE. Children with SE, even if successfully treated in the ED, should be admitted to the hospital for monitoring and observation. Rarely, a child may enter the ED in absence status. In this case, the child may be sitting in a confused or dreamy state. Such attacks may last for hours or even days. The drug of choice in the treatment of absence status is a benzodiazepine at the dosages outlined above.
At times, a child may present with continual focal seizure activity (with or without clouding of consciousness), a condition known as epilepsia partialis continua. The treatment for partial seizures is less urgent than that for generalized seizures, and such seizures are often intractable to anticonvulsant medication. In such cases, fosphenytoin in a dose of 15 to 20 mg per kg can be infused slowly. All such patients should be admitted to the hospital for further observation and evaluation. Other pharmacologic attempts to control these focal seizures should be performed in the hospital.
The decision to initiate long-term prophylactic therapy with anticonvulsant medications is based on a consideration of a number of factors, including the patient’s age, type of seizure, risk of recurrence, coexisting medical conditions, and family factors. The consequences of further seizures must be balanced against the potential side effects of the anticonvulsant agents. Treatment is seldom started after a single, uncomplicated nonfebrile seizure because most such patients will not experience a seizure recurrence. It is preferable to make long-term treatment decisions in conjunction with the provider who will be responsible for ongoing follow-up of the patient, either a neurologist or the child’s primary care physician. Sometimes, it may be necessary to begin prophylactic treatment in the ED, pending a more complete outpatient evaluation.
Disposition
Hospital admission is generally required for children who have had a prolonged seizure requiring acute treatment with anticonvulsant medication. With the exception of very young infants, other children, even those with a first-time seizure, can generally be followed as outpatients if they appear well after the seizure, follow-up can be ensured, and the parents are comfortable with home management. Seizure first aid should be explained to the family before discharge. Some practitioners may choose to prescribe rectal diazepam as a rescue medication until a decision is made about instituting chronic anticonvulsant therapy.
After a simple febrile seizure, hospitalization is seldom necessary, and children may be followed by their primary physician. Some useful information can be given to parents after a first febrile seizure. First, they should be informed of the benign nature of the convulsions and the lack of evidence that they cause any type of neurologic injury. Approximately one-third of children with a first febrile seizure will have another one. Of recurrences, 75% occur within 1 year, and they are uncommon beyond 2 years; fewer than 10% of children with febrile seizures have more than three. The recurrence rate is lower if the seizures begin after the first year of life, and the risk is also reduced in children with higher temperature and longer duration of fever before the initial febrile seizure. For example, the recurrence risk is about 35% when the first seizure occurs at a temperature of 38.5°C (101.3°F), compared with a risk of 13% with a temperature of 40°C (104°F). Having a complex first febrile seizure (even febrile SE) does not increase the risk of recurrence, nor does it increase the chance that a recurrent seizure, if it occurs, will be complex.
Many parents are concerned that febrile seizures will lead to future epilepsy. A child who has had a febrile seizure but no other risk factors for epilepsy may have a slightly increased risk of future nonfebrile seizures, but the magnitude of this increase is still extremely small: 1% to 2% lifetime risk versus a 0.5% to 1% lifetime risk in the general population. Several risk factors that increase the likelihood of a child experiencing future nonfebrile seizures have been identified. These risk factors include a family history of epilepsy, a complex febrile seizure, and the presence of an underlying neurologic or developmental abnormality. Importantly, even with two or more of these risk factors, the risk of epilepsy is only 10%. Thus, for most children with no risk factors, the parents may be reassured that future epilepsy, although possible, is extremely unlikely. Furthermore, there is no association between febrile seizures and any type of developmental or learning disabilities.
DISORDERS THAT PRESENT WITH HEADACHE (SEE ALSO CHAPTER 54 PAIN: HEADACHE)
Migraine
Goals of Treatment
For children with a known diagnosis of migraine, acute management is aimed at reduction of symptoms (which may include nausea and vomiting as well as pain) to a point where home management is feasible. When a diagnosis of migraine has not been established, the emergency clinician must also evaluate for other potential causes of headache.
CLINICAL PEARLS AND PITFALLS
• “Classic” findings are present in a minority of children with migraine, and clinical characteristics such as duration and laterality tend to differ from those in adult patients.
• There is limited pediatric evidence to support most commonly used acute migraine treatments. Nonsteroidal anti-inflammatory agents and prochlorperazine are the best studied.
• While commonly prescribed, opioids are not recommended as first-line treatment for migraine.
Current Evidence
Migraine—recurrent headaches separated by long, symptom-free intervals—is probably the most common specific cause of episodic headaches in afebrile children. In epidemiologic studies, prevalence estimates for migraine in children range from 3% to 10%. A number of forms of migraine are recognized. Migraine is considered classic when the headache is well localized and preceded by an aura and considered common when it is not. The common form of migraine predominates in children. Cluster headaches, which are unilateral, occur in runs and are associated with autonomic changes. They represent a rare migraine variant in childhood. Cyclic vomiting, a syndrome of recurrent, discrete attacks of abdominal pain, nausea, vomiting, and pallor, is also believed to be a migraine variant, sometimes called abdominal migraine.
Migraine is a result of an underlying hyperexcitable cerebral cortex. In a genetically predisposed individual, a variety of stimuli can trigger episodes of “cortical spreading depression,” a slowly propagating wave of neuronal hyperpolarization followed by depolarization. This in turn triggers a neuronally mediated vascular instability that results in intracranial hypoperfusion (which may produce the migraine aura of premonitory motor, visual, or sensory symptoms), followed by vasodilation and a sterile, neurogenic inflammation, which are responsible for the headache.
Clinical Considerations
Clinical Recognition. Prolonged (up to 24 to 48 hours), moderate to severe headache is characteristic of migraine. The headaches may be pulsating and unilateral but this pattern is less common in children than in adults. Migraine is commonly associated with nausea, vomiting, abdominal pain, and photophobia or phonophobia. Auras occur in less than half of children. Occasionally, the attacks awaken the children from sleep.
A family history of migraine is helpful in diagnosis, and a disproportionate number of children who experience migraines have episodes of motion sickness, dizziness, vertigo, or frank paroxysmal events.
Initial Evaluation. The diagnosis of migraine is based almost exclusively on the history and is supported by the absence of abnormalities on examination. There are no diagnostic laboratory tests or imaging studies. The physical examination usually shows no focal neurologic deficits, although hemiplegia and ophthalmoplegia may occur in complicated migraine.
Common trigger factors for migraine in children include emotional stress, lighting changes, and minor head trauma. Particularly in adolescents, it is useful to screen for depression or other psychosocial stressors that may warrant separate treatment. Nitrates (e.g., lunch meats) and tyramine (cheeses) are less common but important food triggers.
Given an accurate history, differentiation from tension headaches, sinusitis, and headaches secondary to intracranial lesions is usually possible; studies such as EEG, CT, and MRI are rarely indicated. In children with focal neurologic deficit and no prior history of such episodes due to migraine, urgent neuroimaging should be considered.
Initial evaluation should include a determination of the level of pain using a validated scale. For patients with known migraine, establishing a target pain level for acute treatment is also helpful.
Management. A number of agents are available for the treatment of acute migraine (Table 105.4). For many children, mild oral analgesics such as acetaminophen or ibuprofen combined with bed rest may provide sufficient relief and should be considered the first-line agents of choice. Ketorolac (Toradol), a nonsteroidal anti-inflammatory agent for parenteral use, may be used when nausea or vomiting limits oral intake. A short course of a narcotic analgesic such as oxycodone may rarely be needed if nonnarcotic agents have failed, especially if the headache prevents sleep.
Dopamine receptor antagonists such as metoclopramide (Reglan), prochlorperazine (Compazine), and promethazine (Phenergan) are commonly used in the ED setting, especially in the presence of nausea and vomiting. These agents have the potential to produce dystonic reactions; it is common to prophylactically coadminister diphenhydramine. Because they have fewer side effects, ondansetron (Zofran) and granisetron (Kytril) have also become first-line agents in the treatment of nausea and vomiting associated with migraine headaches, although these medications have not been studied as primary treatment of migraine headaches.
TABLE 105.4
AGENTS FOR ACUTE TREATMENT OF MIGRAINE
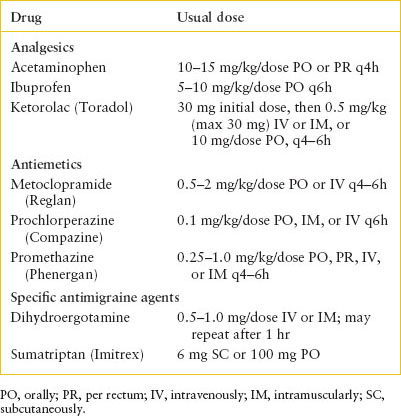
Sumatriptan succinate (Imitrex) is a serotonergic agent available for oral, intranasal, or subcutaneous administration. Its effectiveness in relieving symptoms of acute migraine has been demonstrated in clinical trials in children and adults, but it has not been approved by the U.S. Food and Drug Administration for use in younger children. The dose for children 12 years and older is 6 mg subcutaneously or 100 mg orally. Sumatriptan is generally well tolerated; side effects include irritation at the injection site, flushing, tachycardia, disorientation, and chest tightness that last for several minutes after parenteral administration. In one trial, adverse effects were more common in younger children. A reasonable approach is to use sumatriptan after a trial of analgesics in an older child, although older children or adolescents with recurrent migraine and a history of successful treatment with sumatriptan in the past may benefit from earlier use of this agent. Other agents in this family include rizatriptan (Maxalt), almotriptan (Axert), and zolmitriptan (Zomig). Triptans should not be administered to children with complicated migraines.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


