NEONATAL EMERGENCIES
NICKIE NIFORATOS ANDESCAVAGE, MD, DEENA BERKOWITZ, MD, MPH, AND LAMIA SOGHIER, MD, FAAP
INTRODUCTION AND INITIAL ASSESSMENT
Goals of Emergency Care
The neonate is in a fragile state of transition to extrauterine life. Newborns have limited ability to maintain temperature and glucose and they have limited cardiopulmonary reserves to compensate for dehydration, sepsis, or other stressors. Newborns may exhibit apnea, rather than tachypnea, in response to hypoxia. They are immunocompromised and are unable to communicate subjective findings. Every clinical finding is likely to be more subtle in the neonate than in the older infant. Therefore, the goals of emergency care are to triage and treat neonates urgently until serious disease has been ruled out, while maintaining body temperature and serum glucose.
KEY POINTS
 Newborns have very little reserve to compensate when acute illnesses occur.
Newborns have very little reserve to compensate when acute illnesses occur.
 Tachypnea may be the only presenting sign of congestive heart failure.
Tachypnea may be the only presenting sign of congestive heart failure.
 Hypothermia, rather than fever, may be a neonate’s response to sepsis.
Hypothermia, rather than fever, may be a neonate’s response to sepsis.
 Weight loss is the most sensitive sign of dehydration in the newborn. A loss greater than 10% of birth weight during the first 10 to 14 days of life should be thoroughly investigated.
Weight loss is the most sensitive sign of dehydration in the newborn. A loss greater than 10% of birth weight during the first 10 to 14 days of life should be thoroughly investigated.
 Careful attention must be paid to maintenance of temperature and glucose in the ED.
Careful attention must be paid to maintenance of temperature and glucose in the ED.
RELATED CHAPTERS
Signs and Symptoms
• Respiratory Distress: Chapter 66
• Septic Appearing Infant: Chapter 68
Medical, Surgical, and Trauma Emergencies
• Cardiac Emergencies: Chapter 94
Current Evidence
Evaluation of the newborn poses many unique challenges to clinicians. The perinatal transition from fetal to extrauterine life requires a number of critical, coordinated changes in cardiopulmonary physiology that can take several hours to several days to be completed. Many neonatal conditions are detected within the first few days of life; however, neonates often present to the emergency department (ED) with critical conditions that manifest at home after routine discharge. For example, certain congenital anomalies may be life-threatening, and yet may not present until after this transition has been completed and the newborn has been discharged home. Other perinatally acquired conditions may not present until days later due to their insidious onset. Additional challenges include subtle neonatal presentations and more severe symptoms in response to pathogens because of immature immune function and lack of energy stores. The immune system is entirely dependent on passive immunity provided from the mother during pregnancy, leaving the neonate immunocompromised and susceptible to life-threatening infections. Furthermore, infants have very little in terms of cardiopulmonary reserve, so that the sick infant can go from well appearing to critically ill and cardiopulmonary arrest in a short period of time. Finally, pathogens to which neonates are likely to be exposed during birth include aggressive bacteria and viruses, including Group B streptococcus, gram-negatives, and herpes simplex.
Regardless of the etiology of injury or illness, neonates have a very limited ability to communicate critical changes in health. Infants cannot express subjective data, and often new parents are unable to identify critical changes in infant behavior. As such, it is important for the caregiver to gather and interpret subtle changes in vital signs and the physical examination to avoid catastrophic injury to the ill newborn.
In this chapter, we will highlight the major differences between neonatal anatomy and physiology compared to that of older children. We will also provide clinicians with a concise synopsis of common neonatal disorders that may be encountered in the ED.
Goals of Treatment
Given the unique challenges of neonatal care, the primary goals of treatment are twofold: (1) to distinguish early signs of a sick infant from normal newborn behaviors and (2) to provide timely intervention to prevent permanent injury or death in the case of an ill neonate.
Clinical Considerations
Clinical Recognition
To assist in the clinical recognition of a sick infant, close attention must be paid to the vital signs obtained in triage. Subtle changes in vital signs can often be the only indication of serious illness, and the early detection of these changes can alert the clinician to intervene prior to the loss of physiologic reserve and cardiopulmonary collapse.
Weight
Each newborn encounter should include a weight check. This should be compared to weight at birth. The Centers for Disease Control (CDC) growth charts provide normative values for weight and length in boys and girls. Birth weight below the 10th percentile identifies the small for gestational age (SGA) and that above the 90th percentile identifies the large for gestational age (LGA) infant. Both SGA and LGA infants are at risk for physiologic disturbances. SGA infants have decreased fat stores, which can leave the infant more susceptible to hypoglycemia and electrolyte disturbances. Additionally, SGA infants are much more sensitive to environmental changes and cannot thermoregulate as well as older infants. Hypothermia in the SGA infant can depress the autonomic nervous system which may result in bradycardia and hypotension. The LGA infant is commonly born to a diabetic mother. The high levels of growth hormone and insulin and lack of sufficient postnatal glucose delivery result in hypoglycemia and electrolyte disturbances, such as hypocalcemia. LGA infants are also at risk for polycythemia. Severe polycythemia can compromise cardiovascular function, as well as increase the risk of hyperbilirubinemia.
Normal weight patterns include a brief period of weight loss in the immediate postnatal period, followed by regular, consistent weight gain for the first few months of life. Weight loss is the most sensitive sign for dehydration in the newborn, particularly as other common signs, such as decreased urine output or skin turgor are not reliable findings in this group. A loss of more than 10% of birth weight is cause for concern, and should be evaluated, particularly for dehydration, hypoglycemia, and electrolyte disturbances. Neonates that have not regained birth weight by 14 days of life should also be evaluated thoroughly. Most commonly, slow weight gain is due to decreased milk transfer in breast-fed infants, but can also signify increased caloric and metabolic demands due to underlying congenital anomalies of the cardiovascular, respiratory, gastrointestinal, or renal systems. Neurologic diseases, particularly those with decreased tone and motor strength, may also present with weight loss or poor weight gain if the infants do not have the strength to adequately suck and swallow. A basic metabolic panel (BMP) should be checked for signs of hypernatremia or other electrolyte disturbances. Neonates who are receiving suboptimal feeds due to inadequate supply may need to be supplemented with formula. Referral of the infant to the pediatrician for serial follow-up is recommended. Alternately, those requiring assistance with poor breast-feeding technique may be supplemented with expressed breast milk or formula and referred to a lactation consultant.
Heart Rate
Normal resting heart rate for a neonate can range between 80 and 180 beats per minute. Over the first few weeks of life heart rate will decrease to 80 to 140 beats per minute. During rest and deep sleep, the heart rate will typically be in the lower range, and in the higher range with activity or agitation. A healthy infant will show variability in heart rate with subtle changes during inspiration and exhalation and when alternating between sleep and alert states. Loss of heart rate variability has been associated with systemic illnesses, such as shock and infection.
Bradycardia can be defined as either a 20 to 30 beat decrease below the infant’s baseline or below 80 beats per minute at rest. Neonates respond to poor cardiac output by increasing their heart rate because stroke volume cannot increase acutely. Therefore, bradycardia is often a late sign of cardiac failure after the normal compensatory mechanisms have collapsed. Sinus bradycardia may also occur with hypothermia, hypothyroidism, malnutrition, or electrolyte disturbances. Premature infants may also present with recurrent bradycardic events, particularly during episodes of increased vagal tone due to gastrointestinal reflux, emesis, or apnea. A 12-lead electrocardiogram can help distinguish sinus bradycardia from conduction abnormalities and heart disease. Some full-term infants have a low resting heart rate that may reach 80 to 90 beats per minute. Any infant with bradycardia who shows other signs of cardiovascular instability, such as hypotension and poor capillary refill, or systemic signs of lethargy or poor feeding, should be treated and stabilized immediately.
Neonatal tachycardia can be defined as a heart rate >190 beat per minute. Sinus tachycardia may occur in the setting of shock, dehydration, or hypovolemia, as a mechanism to increase cardiac output. Neonatal tachycardia can also occur with fever, hyperthyroidism, severe anemia, hypoglycemia, or electrolyte disturbances, such as hypocalcemia. The most common conduction abnormality that results in tachycardia in the neonate is supraventricular tachycardia (SVT), which can lead to cardiac failure and shock if sustained.
For more details on clinical considerations and management of heart rate anomalies in the neonate, see Section: Neonatal Cardiac Emergencies.
Respiratory Rate
The normal respiratory rate of a newborn is between 30 and 60 breaths per minute. Periodic breathing is a normal finding that presents with pauses between breaths that can last up to 10 seconds. Most commonly, the infant will have 2 to 3 pauses close together, followed by a series of short, shallow breaths. With periodic breathing, the infant will remain well appearing and without signs of respiratory distress. In contrast, apneic spells last greater than 20 seconds OR are associated with cyanosis or bradycardia. Apneic events are never a normal finding, and should be further evaluated (see Chapter 9 Apnea).
Tachypnea can result from systemic changes, such as fever, hyperthyroidism, metabolic acidosis, cardiac disease, or pulmonary disease. It is often the only presenting sign of congestive heart failure. Tachypnea can be a presenting sign for a number of metabolic disorders, particularly urea cycle defects, as well as renal disorders, where the body is compensating for metabolic acidosis. Tachypnea is common with respiratory illnesses, such as bronchiolitis or pneumonia, but also in premature infants with chronic lung disease, or infants with a history of congenital pulmonary lesions such as congenital diaphragmatic hernia. The underlying disease causing tachypnea should be addressed prior to the development of hypoxia and respiratory failure.
For more details on clinical considerations and management of respiratory rate anomalies in the neonate, see Section: Neonatal Respiratory and Airway Problems.
Pulse Oximetry
At sea level, normal pulse oximetry saturation values are greater than 94%; 89% and higher are acceptable at higher elevations, such as in Denver and Salt Lake City. Pulse oximetry saturation (POS) values in healthy newborns normalize within the first few hours of life, with a mean value 97% by 24 hours. POS values increase incrementally with gestational age, and show variability with infant state. Crying neonates are more likely to have lower values compared to quiet or sleeping infants.
Pulse oximetry should be used for any infant with tachypnea, dyspnea, or any signs of cardiorespiratory illness. Cyanosis may be absent in the face of significant hypoxemia if the hemoglobin is low (see Section: Color Changes). It is important to note that acrocyanosis is a common neonatal finding in healthy infants that can persist for several days to weeks, particularly in cool environments. This is in contrast to infants with central cyanosis, which should always be evaluated with pulse oximetry and arterial blood gas measures of oxygenation.
Hypoxia is never normal in the term newborn, and may be due to respiratory or cardiac anomalies. Increasingly, pulse oximetry has been used as a screening method for critical congenital heart diseases (CHDs) that produce hypoxemia. CHD screening should be completed after 24 hours of life to decrease the incidence of false positives, but within the first week. Importantly, the pulse oximetry screen for CHD does not detect nonhypoxic heart disease (e.g., coarctation of the aorta, single ventricle, or double outlet right ventricle). The American Academy of Pediatrics (AAP) and the CDC endorse screening for critical CHD for all newborns (Fig. 104.1). An infant who fails the initial screen should be referred for an echocardiogram.
Hypoxia without respiratory distress, specifically without signs of grunting, retractions, or accessory muscle use, is more common in cardiac disease than respiratory disease. To better distinguish the etiology of hypoxemia, the clinician can perform the hyperoxia test. To complete the test, the infant is given 100% oxygen to breathe for 5 to 10 minutes. Serial pulse oximetry or arterial blood gas measurements are obtained on room air and after the infant has breathed 100% oxygen. If there is little to no increase in oxygenation, the hypoxia can be attributed to extrapulmonary causes of right-to-left shunting. Extrapulmonary right-to-left shunting occurs in persistent pulmonary hypertension and in cardiac disease. To distinguish between the two, the clinician can perform the hyperventilation test. In this circumstance, hyperventilating to a PaCO2 of 25 to 30 mm Hg in conjunction with 100% oxygen is more likely to elicit an increase in PaO2 levels (typically >100 mm Hg) in persistent pulmonary hypertension of the newborn (PPHN) due to relaxation of the pulmonary bed. Infants that continue to have low PaO2 despite hyperoxia and hyperventilation are more likely to have a fixed, intracardiac right-to-left shunting. In either circumstance, an echocardiogram is the definitive study to differentiate between the two.
Supplemental oxygen should be administered to infants with suspected or confirmed respiratory disease. Hypoxic premature infants with lung disease should be administered oxygen judiciously, particularly if they have not corrected to term gestation. Hyperoxia is associated with retinopathy of prematurity, increased risk of pneumonia, and exacerbations of chronic lung disease. Target saturations for this population should be discussed with a neonatologist. Similarly, oxygen administration to infants with CHD can alter pulmonary vascular resistance and influence the direction and degree of intracardiac shunts, potentially leading to congestive heart failure. Target saturations for this population should be discussed with a cardiologist.
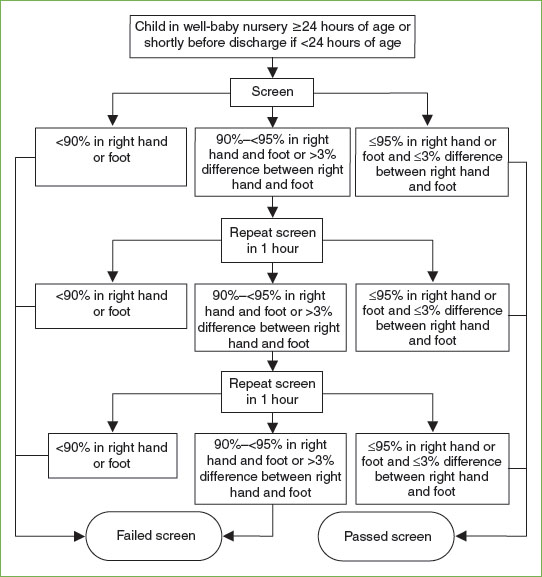
FIGURE 104.1 Congenital heart disease screening algorithm. (From Kemper AR, Mahle WT, Martin GR, et al. Strategies for implementing screening for critical congenital heart disease. Pediatrics 2011;128(5):e1259–1267.)
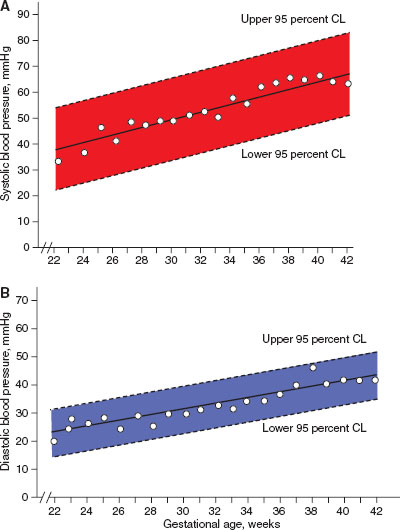
FIGURE 104.2 Neonatal blood pressure norms based on gestational age. A: Systolic blood pressure norms. B: Diastolic blood pressure norms. (From Zubrow AB, Hulman S, Kushner H, et al. Determinants of blood pressure in infants admitted to neonatal intensive care units: a prospective multicenter study. J Perinatol 1995;15(6):470–479.)
It is important to note that pulse oximetry measures are dependent on adequate pulse pressure, and so any low perfusion state may lead to falsely low pulse oximetry readings. Pulse oximetry is also unable to detect significant hyperoxia or severe hypoxemia. The presence of other hemoglobin forms, such as methemoglobin or carboxyhemoglobin, may not be detected by pulse oximetry. In any of these circumstances, arterial blood sampling for PaO2 and cooximetry for carboxyhemoglobin or methemoglobin may be needed to better understand the infant’s respiratory physiology.
Blood Pressure
Blood pressure monitoring in the newborn requires specific equipment and interpretation. Most commonly, indirect blood pressure monitoring utilizes an occlusive cuff device that functions identically to pediatric and adult cuffs. Neonatal blood pressure cuff width should measure ∼40% of the member circumference. A cuff that is too loose can result in inaccurate measurement of blood pressure. To increase accuracy, the cuff should be placed at the same level as the heart, typically in the upper extremity. Normal ranges for blood pressure increase within the first few hours to days of life and are dependent on the infant’s weight and gestational age at birth, and should be interpreted accordingly (Figs. 104.2 and 104.3).
Hypertension is confirmed after serial accurate measurements reveal consistent elevations above 95% for age and weight. The most common causes of neonatal hypertension include umbilical artery catheterization, renovascular disease, parenchymal renal disease, and chronic lung disease of prematurity. Additionally, coarctation of the aorta, hyperthyroidism, congenital adrenal hyperplasia, and increased intracranial pressure can cause neonatal hypertension and can be life-threatening if left untreated. Most infants with hypertension are asymptomatic. When symptoms are present, they are often nonspecific (lethargy, poor feeding, apnea) and do not necessarily correlate with the degree of hypertension. Initial evaluation should include blood pressure measurement in all four extremities, urinalysis, urine culture, blood urea nitrogen, serum creatinine, electrolytes, and calcium. It is important to note that the absence or presence of hematuria, proteinuria, or azotemia vary in this age group and cannot be used in isolation to diagnose renovascular disease. If the history and physical examination are suggestive of endocrine, neurologic, or intoxication causes of hypertension, additional testing may be needed. Renal ultrasonography with Doppler evaluation should also be included to evaluate for renovascular and parenchymal disease. Echocardiography should be considered to assess left ventricular function. Determining when to institute pharmacotherapy for hypertension is based on the underlying etiology, severity of hypertension, and presence of symptoms. The decision to initiate therapy should be done in consultation with pediatric nephrologist.
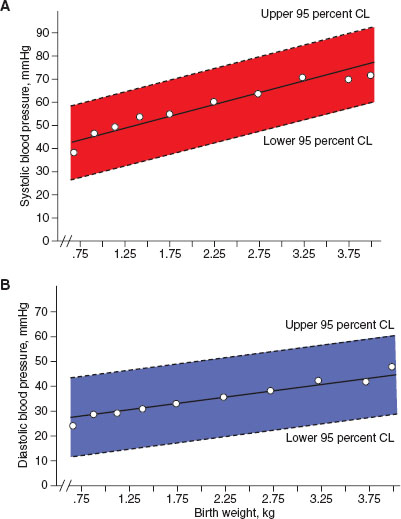
FIGURE 104.3 Neonatal blood pressure norms based on birth weight. A: Systolic blood pressure norms. B: Diastolic blood pressure norms. (From Zubrow AB, Hulman S, Kushner H, et al. Determinants of blood pressure in infants admitted to neonatal intensive care units: a prospective multicenter study. J Perinatol 1995;15(6):470–479.)
Hypotension in a neonate can result from volume depletion, hemorrhage, sepsis, or cardiac failure. Detecting hypotension in the preterm or SGA infant can be challenging, as noninvasive monitoring can routinely overestimate blood pressure values. It is imperative to treat hypotension aggressively to prevent end-organ damage and multisystem organ failure. First-line treatment is to provide intravascular replacement with iso-osmotic fluids, typically normal saline, or packed red blood cells in the setting of acute hemorrhage. Typical resuscitation volumes are 10 mL per kg over 30 minutes, with more judicious use in the premature infant. Excessive volume expansion in the preterm neonate is associated with higher morbidity; therefore early administration of pressors is necessary if there is a limited response to volume. Treatment of hypotension should be directed at improving perfusion and cardiac function, rather than aiming for a desired blood pressure value. This is of particular importance in conditions that widen the pulse pressure, where systolic pressures are adequate but the mean arterial pressure underestimates perfusion pressure.
Temperature
Rectal thermometry is considered the reference standard for measurement of body temperature in neonates. However, it is important to note that mechanical trauma from rectal thermometry in a newborn can result in peritonitis and abscess formation, and should be performed with caution. It is also contraindicated in patients with neutropenia. Similarly, infants receiving active intervention for temperature control require continuous thermometry that is better accomplished with electronic axillary thermometry. Temperature readings may vary according to the site measured so that reference ranges should be interpreted with its specific set of normal values. In general, hypothermia in a neonate occurs when the temperature is less than 36.5°C, and fever occurs when temperatures exceed 38°C, although the threshold for associated metabolic changes that occur with each can vary from infant to infant.
Infants have a very large ratio of surface area to body mass, low fat stores, and immature thermoregulatory centers, all of which leave them at increased risk for cold stress. Neonates naturally respond to cold stress by becoming hypermetabolic, vasoconstricted, hyperactive, tachycardic, tachypneic, and acidotic. Heat loss after a week of life commonly occurs through radiation, and is greatly influenced by ambient temperature, humidity, and the temperature of surfaces to which the infant is exposed. Therefore, whenever possible, it is important for the clinician to minimize exposure to cold air and surfaces during examination or observation by placing the baby under an open radiant heater.
Hyperthermia or fever is most often noted as a sign of hypermetabolism in a septic infant. However, hypothermia can also be a subtle sign of sepsis, due to a markedly diminished response to bacterial pyrogens in neonates (see Chapter 87 Fever in Infants).
Temperature regulation can also be altered in infants with hypoxic–ischemic injury. Therapeutic hypothermia decreases neurologic morbidity if instituted soon after delivery for infants with perinatal injury (see Section: Neonatal Neurological Emergencies). Regardless of the timing or etiology of hypoxia–ischemia, fever should be aggressively treated in this population as hyperthermia can worsen neurologic injury.
COLOR CHANGES AND DERMATOLOGIC FINDINGS
Goals of Emergency Care
The goal is to recognize the difference between benign presentations, such as acrocyanosis, and life-threatening conditions, such as true cyanosis and pallor.
KEY POINTS
 Evaluation of jaundice requires assessment of both direct and indirect bilirubin.
Evaluation of jaundice requires assessment of both direct and indirect bilirubin.
 Acceptable levels of indirect hyperbilirubinemia depend on both prematurity and postnatal age.
Acceptable levels of indirect hyperbilirubinemia depend on both prematurity and postnatal age.
 Many neonates with herpes simplex virus (HSV) infection have a negative maternal history of HSV.
Many neonates with herpes simplex virus (HSV) infection have a negative maternal history of HSV.
RELATED CHAPTERS
Signs and Symptoms
• Jaundice: Conjugated Hyperbilirubinemia and Jaundice: Unconjugated Hyperbilirubinemia: Chapters 39 and 40
• Septic Appearing Infant: Chapter 68
Medical, Surgical, and Trauma Emergencies
• Cardiac Emergencies: Chapter 94
Color Changes
Cyanosis
CLINICAL PEARLS AND PITFALLS
• Cyanosis may not be visible toward the end of the neonatal period because of physiologic anemia.
• Benign acrocyanosis may involve the perioral region but spares the lips and mucus membranes.
• Diarrheal illness and/or dehydration can lead to acquired methemoglobinemia.
Current Evidence. Cyanosis refers to a blue tone visible in the skin and mucus membranes, caused by desaturated or abnormal hemoglobin. The human eye can detect cyanosis when there is at least 5 gm/dL of reduced hemoglobin. Therefore hemoglobin oxygen desaturation may be missed if there is anemia. Conversely, abnormal hemoglobin may be saturated with oxygen yet unable to release to the tissues, resulting in visible cyanosis. Methemoglobinemia is a classic example of this situation. Central cyanosis is caused by deoxygenated blood entering the systemic circulation. This is usually due to CHD, specifically cardiac defects allowing systemic venous blood to bypass the lungs (right-to-left shunt), but central cyanosis may also be caused by respiratory compromise or pharmacologic agents. Acrocyanosis, the transient blue discoloration of the hands and feet in response to vasomotor instability or a cool environment, is caused by vasoconstriction of the small arterioles and does not reflect reduced systemic arterial oxygenation. Mottling is the patchy-colored appearance of the body surface, resulting from dilation of the superficial veins showing through the thin neonatal skin.
Goals of Treatment. The goals of ED evaluation of the cyanotic infant include early recognition of cardiorespiratory pathology or pharmacologic causes.
Clinical Considerations
Clinical recognition. Proper lighting is important to assess cyanosis in neonates. Location of cyanosis helps determine its cause. Cyanosis noted in the mucus membranes, tongue, trunk, and extremities is central. In contrast, acrocyanosis is limited to hands, feet, and perioral region, with the tongue and rest of skin remaining pink. This condition may be associated with cool ambient temperature. Acrocyanosis is benign and may resolve with warming. Local blue discoloration of a single extremity could be the result of compromised distal circulation. A local blue hue to skin may also be the result of pigment from blue clothing dye.
Triage consideration. “Blue babies” should be evaluated promptly for cardiorespiratory disease. The degree of oxygen desaturation associated with cyanosis should be documented by pulse oximetry.
Clinical assessment including labs, x-ray, and management. When obtaining history, relevant questions include the following: When was the color change first noted? Is it persistent or intermittent? For example, choanal atresia will cause cyanosis at rest, which improves with crying, whereas the cyanosis of congenital cardiac disease will often worsen with crying because of increased pulmonary vascular resistance. Does the cyanosis improve with oxygen? Are there other accompanying symptoms? In cyanotic cardiac disease (i.e., right-to-left shunt), the breath sounds will be normal with symmetric chest excursion. In contrast an infant with cyanosis due to pulmonary disease or congestive heart failure will have wheezes or crackles and accessory chest muscle use. With pulmonary disease or congestive heart failure, the increase in saturation may be dramatic when the infant receives increased oxygen (see hyperoxia test, under Section: Neonatal Cardiac Emergencies).
Cyanosis accompanied by mottling in a lethargic neonate with tachycardia indicates shock. Sepsis, hypovolemia, intra-abdominal surgical emergency, and metabolic crisis from inborn errors of metabolism (IEM) should be considered in addition to primary cardiorespiratory problems. Early recognition and volume resuscitation is critical for treatment.
Methemoglobinemia is characterized by a cyanotic infant without underlying cardiac or pulmonary disease. The infant can look cyanotic to gray, with an almost normal-appearing pulse oximetry of 85% or greater. Supplemental oxygen will not alter the color. Methemoglobinemia is confirmed by venous or capillary blood gas or the persistence of a chocolate-brown color of a blood drop on filter paper. Initial treatment is first searching and removing the offending agent, which is most often topical anesthetic agents, aniline dyes, and high levels of nitrate in the water supplies. Levels of methemoglobinemia above 20% are associated with clinical symptoms. If methemoglobinemia is greater than 30% of total hemoglobin, consider a dose of methylene blue, 1 to 2 mg per kg, given over 5 minutes.
Jaundice
CLINICAL PEARLS AND PITFALLS
• Jaundice within the first 24 hours of life is pathologic.
• Evaluation of jaundice requires assessment of both direct and indirect bilirubin.
• Acceptable levels of indirect hyperbilirubinemia depend on both prematurity and postnatal age.
• Acute bilirubin toxicity results in increase lethargy and progressive encephalopathy in the neonate. Untreated, persistent bilirubin encephalopathy results in kernicterus, a permanent brain injury.
• Galactosemia should be considered in infants who have jaundice that persists beyond 3 weeks of age.
Current Evidence. Jaundice is a yellow appearance of the skin or sclera caused by elevated bilirubin levels. Bilirubin accumulates with excessive hemolysis, failure of hemoglobin to conjugate with glucuronic acid in the liver, or inadequate excretion through the liver canaliculi or bile ducts. Unconjugated bilirubin is reported as indirect, and indicates excessive red blood cell hemolysis or inability of the liver to keep pace with conjugation of bilirubin produced by hemolysis. Conjugated bilirubin is reported as direct and is elevated because of obstructed excretion.
Goals of Treatment. The goals of ED evaluation of the icteric infant include rapid diagnosis of the acutely treatable causes of icterus (sepsis, obstruction, metabolic disease), prevention of kernicterus and brain injury, and reassurance when the jaundice is physiologic.
Clinical Considerations
Clinical recognition. In a child with true jaundice, the sclera will be yellow. Jaundice in the first 24 hours of life of a term newborn is pathologic. Full-term, well-appearing neonates over 1 day of age often have physiologic or breast-milk jaundice. Physiologic jaundice is benign. Bilirubin levels spike close to day 3 of life and then decrease. Breast-feeding jaundice may present in the first few days before sufficient milk production or later on for unknown reasons. Breast-fed infants may have prolonged unconjugated hyperbilirubinemia lasting up to several weeks thought to be related to compounds in breast-milk.
Clinical assessment. Infants presenting with jaundice require a careful history, looking for timing of jaundice onset, stool and urine color, breast or bottle feeding, maternal and infant blood type, traumatic delivery, and maternal history of diabetes, hepatitis, and medications. A careful examination should assess for cephalohematoma and signs of infection. Acholic stools generally indicate biliary obstruction or severe hepatic failure.
CBC, total bilirubin level, and direct bilirubin level should be obtained. A direct Coombs test should be obtained if maternal blood type in unknown, hemoglobin is low, or total bilirubin is at levels requiring intervention (see below). Consider blood typing mother and neonate for ABO and Rh factors if unknown. Rh incompatibility is uncommon in mothers who have received prenatal care.
Infants presenting with elevated indirect bilirubin, direct bilirubin less than 15% of the total, and normal stool color have unconjugated hyperbilirubinemia. Causes of unconjugated hyperbilirubinemia include physiologic jaundice, breast-milk jaundice, breast-feeding jaundice, hemolytic disease, blood group incompatibility, infection, dehydration, polycythemia, abnormalities in the conjugating enzyme (UDP-glucuronosyltransferase or UGT) as with Crigler–Najjar syndrome or Lucey–Driscoll syndrome, Gilbert syndrome, or hypothyroidism.
Infants with elevated direct bilirubin have conjugated hyperbilirubinemia, which is always pathologic. Elevated direct bilirubin indicates obstructed excretion, or cholestasis, usually due to biliary obstruction, hepatocellular pathology, or metabolic disorder. Hepatitis, and in particular, idiopathic neonatal hepatitis, is a common cause of conjugated hyperbilirubinemia. This must be distinguished from infectious hepatitis, including hepatitis B, rubella, cytomegalovirus, toxoplasmosis, coxsackie virus, echovirus, HSV, syphilis, Listeria, and tuberculosis. In addition, systemic infectious disease can also result in a toxic hepatitis, particularly systemic infections due to Escherichia coli, Pneumococcus, Proteus, and Salmonella. Prolonged parenteral alimentation is another common cause of conjugated hyperbilirubinemia that can persist several months beyond the cessation of parenteral nutrition. Metabolic disorders of the liver that can also result in conjugated hyperbilirubinemia include alpha 1-antitrypsin deficiency, galactosemia, tyrosinemia, fructosemia, glycogen storage diseases, lipid storage diseases, cerebrohepatorenal syndrome, trisomy 18, cystic fibrosis, hemochromatosis, and idiopathic hypopituitarism.
Obtain CBC, blood culture, albumin, LFT’s PT/PTT, hepatitis serologies, urinalysis, urine culture, and urine-reducing substances. Biliary atresia and neonatal hepatitis are the most common etiologies and patients with biliary atresia have better outcomes the earlier they are diagnosed. Neonates presenting with elevated direct bilirubin with microcephaly, seizures, organomegaly, or petechiae should be evaluated for congenital TORCH (toxoplasmosis, other infections, rubella, CMV, herpes simplex) infections.
Management. Determine if the infant has indirect or direct hyperbilirubinemia. Infants with direct (conjugated) hyperbilirubinemia or acholic stools require admission for further diagnostic evaluation. Treatment of neonatal hepatitis is supportive, with supplementation of the fat-soluble vitamins. Prevention of hepatitis B infection requires early administration of HBIG and HBV vaccines to prevent chronic infection in exposed newborns. Septic infants should be treated with broad-spectrum antibiotics, typically with rapid normalization of hyperbilirubinemia. While some of the metabolic disorders (galactosemia, tyrosinemia, fructosemia, and the storage diseases) may be palliated with dietary restrictions, most progress to chronic cirrhosis and liver failure.
Indirect Hyperbilirubinemia. Infants with physiologic jaundice can be discharged if they do not meet criteria for phototherapy or exchange transfusion. The Subcommittee on Hyperbilirubinemia of the AAP publishes guidelines for management of hyperbilirubinemia in healthy term infants according to total bilirubin levels and infants age (see Figure 40.1). Infants with breast-milk jaundice usually require increased breast-feedings, and close follow-up with pediatrician until adequate milk supply is insured. Infants of diabetic mothers, infants with congenital hypothyroidism, or with resorption from large cephalohematomas may have higher levels of icterus than expected physiologically and may require serial bilirubin level checks.
Pallor
CLINICAL PEARLS AND PITFALLS
• Anemia in the neonate is recognized as pallor most commonly when the hemoglobin falls below 10 g/dL.
Current Evidence. True pallor in the neonate can be due to significant anemia, sepsis, shock, or severe chemical/electrolyte imbalance.
Clinical recognition. Anemia in the neonate is recognized as pallor most commonly when the hemoglobin falls below 10 g/dL.
Clinical assessment. ED evaluation of pallor should include a careful history and physical examination to distinguish between anemia, septic/cardiogenic shock, electrolyte imbalance, and genetically determined fair skin coloring. Questions should be asked regarding perinatal history, the baby’s feeding, level of alertness, occurrence of vomiting or diarrhea, fever, and infant’s responsiveness. Careful physical examination should include attention to vital signs, hydration status, and lethargy, and search for cephalohematoma and hepatosplenomegaly. Consider obtaining a CBC.
Dermatologic Findings
CLINICAL PEARLS AND PITFALLS
• As few as one to two vesicles on an erythematous base, most commonly on the scalp and face, may be the only presenting signs of HSV infection, which should be considered even in the absence of maternal signs or history of infection.
• A tuft of coarse dark hair located in midline lumbosacral region may be associated with spina bifida occulta.
Benign Rashes
Various papular rashes may be observed in the healthy newborn. Characteristic body distribution patterns and age at appearance help differentiate these rashes from more worrisome conditions. Diagnosis can be made by physical examination alone and do not require further evaluation or specific treatment. Parents should be reassured these are not worrisome conditions.
Milia are small 1- to 2-mm ivory or yellow papules located primarily on the forehead, nose, and cheeks of newborns. Milia are keratin retention cysts. They will spontaneously rupture and disappear during the first 3 to 4 weeks of life (Fig. 104.4). Miliaria, or neonatal prickly heat, is caused by sweat retention and is characterized by easily ruptured, 1 to 2 mm vesicles located primarily on the face chest and back. Erythema toxicum is a more generalized eruption of small papules or pustules on an erythematous base that may occur anywhere on the body. Usually presenting during the first 3 to 4 days of life, these lesions may be noted as late as 2 weeks of age. If the diagnosis is in question, a smear of the papular contents will show a predominance of eosinophils with no organisms and relative absence of neutrophils (Fig. 104.5). Neonatal acne is characterized by erythematous papules or pustules confined primarily to cheeks, chin, and forehead. Lesions are caused by circulating maternal hormones, usually appear at 3 to 4 weeks of age and disappear within a few weeks (Fig. 104.6). Diaper rash is located on the skin covered by the diaper. Irritants and contact dermatitis are often the initial precursor to the rash. Candida infections are common and diagnosed if the rash is present in the intertriginous folds. Candida infections are beefy red with well-demarcated borders and satellite papules and pustules. Mostly a clinical diagnosis, candida can be confirmed if necessary by microscopic examination looking for budding yeasts and pseudohyphae.
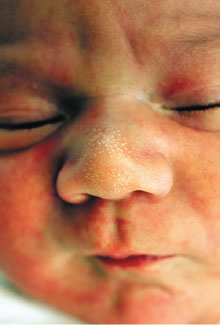
FIGURE 104.4 Milia. (From Jensen S. Nursing health assessment. Philadelphia, PA: Lippincott Williams & Wilkins; 2010.)
Significant Neonatal Rashes (See Chapter 64 Rash: Neonatal)
HSV Infection
HSV is the most important condition to consider when evaluating a vesicular eruption because of its significant associated morbidity and mortality.
Current Evidence. Most neonatal HSV infections are caused by HSV type 2. While transplacental transmission can occur, most neonatal HSV disease is acquired perinatally from infection of the maternal genitourinary tract. Primary maternal infection at the time of delivery is associated with a 40% to 50% chance of transmission but most neonates with HSV are born to mothers who did not provide a history of HSV infection or other identifiable risk factors.
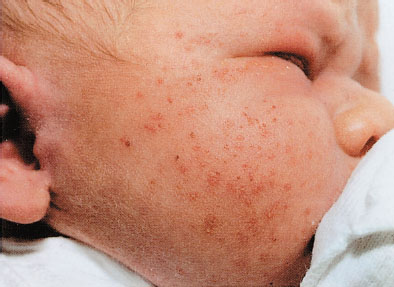
FIGURE 104.5 Erythema toxicum. (From Fletcher M. Phyisical Diagnosis in Neonatology. Philadelphia, PA: Lippincott-Raven Publishers; 1998.)
FIGURE 104.6 Neonatal acne. (Courtesy of Amy Ross, MD. In: Chung EK, Atkinson-McEvoy LR, Boom JA, et al., eds. Visual diagnosis and treatment in pediatrics. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010.)
Clinical Considerations. Goals of treatment: early diagnosis and prompt initiation of antiviral therapy to decrease morbidity and mortality.
Clinical recognition. The infant can appear well at birth but become ill at days 4 to 7, at which time a vesicular eruption may be noted (Fig. 104.7). The most common lesions are small vesicles on an erythematous base which may become pustular in 1 to 2 days. However, early manifestations can be subtle and nonspecific. HSV should be suspected in infants with or without vesicles who present with sepsis, respiratory distress, focal neurologic signs, or signs of hepatitis or liver failure.
Triage consideration. Neonates presenting with new vesicles should be seen promptly.
Clinical assessment. Neonatal HSV is categorized into three main categories: localized skin, eye, and mouth (SEM) disease; central nervous system (CNS) with or without SEM; and disseminated disease, which may involve CNS, SEM, and other organs. Comprehensive laboratory evaluation should occur in infants with suspected HSV, even if only limited SEM involvement is suspected. Obtain CBC, blood chemistry, liver function tests, urinalysis, blood and urine culture, lumbar puncture, HSV polymerase chain reaction (PCR) of the blood, cerebrospinal fluid (CSF) cell count, glucose, protein, HSV PCR, viral culture, surface culture from conjunctiva, mouth, nasopharynx, and rectum—placed in single viral transport media tube—and evaluation for bacterial or metabolic disease as indicated.
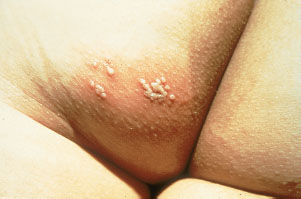
FIGURE 104.7 Herpes simplex infection. (From Goodheart HP, MD. Goodheart’s photoguide of common skin disorders. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2003.)
Management. If a neonate is suspected of having HSV, perform a complete sepsis workup, start acyclovir, and admit the patient to the hospital. Infants may require supportive care with intubation, mechanical ventilation, and cardiac support for disseminated disease. In addition, infants with significant liver dysfunction may require transfusions in the setting of significant coagulopathy and bleeding.
Neonatal Varicella
Current Evidence. Neonatal varicella may develop when maternal varicella infection occurs during last 2 to 3 weeks of pregnancy or the first few days postpartum. The severity of neonatal disease depends on timing of maternal infection. If maternal disease onset is 5 or more days before delivery, the infection in the newborn is usually mild because of transplacental passage of maternal varicella IgG antibodies. If maternal disease is within 4 days before delivery the neonate is at risk of developing severe infection, with mortality as high as 30% caused by pulmonary or visceral involvement. Infants born to mothers who demonstrate chickenpox lesions within 2 to 5 days of delivery should receive varicella zoster immunoglobulin.
Clinical Recognition. The classic rash is described as vesicles on an erythematous base, or “dew drops on a rose petal” (Fig. 104.8).
Goals of Treatment. Early recognition, support, and prompt initiation of antiviral therapy for moderate to severe cases of varicella.
Incontinentia Pigmenti
This is an X-linked dominant disorder with both skin and systemic lesions affecting the eyes, CNS, and skeletal system. The disease presents at birth or shortly after with an inflammatory vesicular or bullous rash that develops in crops over the trunk and extremities. The cutaneous lesions have four phases (inflammatory vesicles or bullae, verrucous lesions, whorled hyperpigmentation, and hypopigmented patches) that may overlap and occur in irregular sequence. Suspected cases should be referred to a dermatologist because of the potential for systemic involvement.
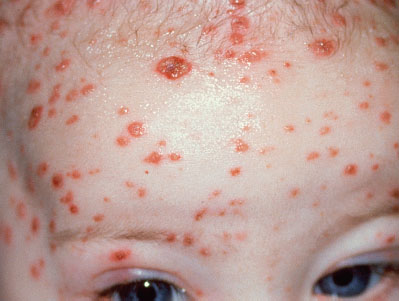
FIGURE 104.8 Varicella infection. (From Burkhart C, Morrell D, Goldsmith LA, Papier A, et al. VisualDx: essential pediatric dermatology. Philadelphia, PA: Lippincott Williams & Wilkins; 2009.)

FIGURE 104.9 Nevus simplex (salmon patch). (Used with permission from Goodheart HP. Goodheart’s photoguide of common skin disorders. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:1.)
Vascular Lesions
Salmon patch (nevus simplex) is the most common vascular lesion of infancy. It is a pale pink macular lesion that is found most commonly on the nape of the neck, forehead, nasolabial region, or upper eyelids. Most resolve by the second year of life. Lesions on the neck may persist for life (Fig. 104.9).
Port-wine stains (nevus flammeus) present at birth as pink to purple macular lesions than can vary tremendously in size (Fig. 104.10). These lesions do not fade with time. Klippel– Trénaunay–Weber syndrome should be considered when the port-wine stain involves a lower limb. Consider Sturge–Weber syndrome when infants present with unilateral facial port-wine stains in a trigeminal nerve distribution. As Sturge–Weber syndrome can involve seizures, intracranial calcifications, and hemiparesis, infants with unilateral facial port-wine stains in trigeminal nerve distribution need further assessment.
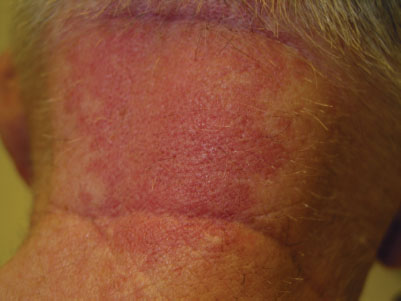
FIGURE 104.10 Nevus flammeus (port-wine stain). (Image provided by Stedman’s.)
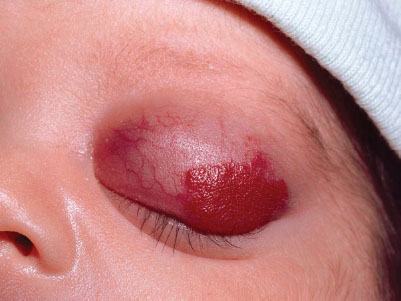
FIGURE 104.11 Capillary hemangioma (strawberry hemangioma). (Courtesy of Dean John Bonsall, MD. In: Chung EK, Boom JA, Datto GA, et al., eds. Visual diagnosis in pediatrics. Philadelphia, PA: Lippincott Williams & Wilkins; 2006.)
Hemangiomas
Capillary hemangiomas (strawberry hemangioma) may be present at birth but usually develop during the first few weeks of life. Lesions may present anywhere on the body, starting as small, well-demarcated telangiectasis or macules that develop into raised scarlet or purple tumors with distinct borders. Most capillary hemangiomas grow rapidly the first 6 months of life, enter into a static period and then recede, usually by 5 years of age (Fig. 104.11). Cavernous hemangiomas are deep-seated capillary hemangiomas that usually present at birth as diffuse swelling with no color change or a bluish hue. Most spontaneously involute over months to years.
Management. Hemangiomas that require intervention in the neonatal period are those that may compromise vital structures, such as the eyes, nares, or auditory canal, or lesions that by their location are susceptible to trauma, ulceration, and secondary infection. Kasabach–Merritt syndrome, which presents as large, rapidly enlarging hemangiomas associated with thrombocytopenia and consumption coagulopathy, should be referred to appropriate specialists for management. Monitor serial CBC and if evidence of significant bleeding/coagulopathy, may require transfusions of red cells, platelets, or coagulation factors.
Pigment Changes
Mongolian spots are poorly circumscribed lesions of bluish-gray maculae generally located over the lumbosacral region, buttocks, and lower limb. Mongolian spots are more commonly noted in infants of African-American, Native American, Hispanic, and East Asian descent. Lesions usually fade during the first year of life and do not require intervention (Fig. 104.12).
Café-au-lait macules are round or oval, brown macular lesions varying in size from less than 1 cm to greater than 20 cm. Though small, infrequent café-au-lait macules are common in the general population, increases in number or size may be a sign of neurocutaneous disease, such as neurofibromatosis. The clinician should carefully examine these infants for other cutaneous or systemic signs of disease (Fig. 104.13).
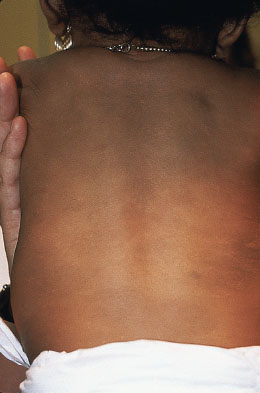
FIGURE 104.12 Mongolian spots. (Courtesy of George A. Datto, III, MD. In: Chung EK, Atkinson-McEvoy LR, Boom JA, et al., eds. In: Visual diagnosis and treatment in pediatrics. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010.)
Ash-leaf macules are irregular hypopigmented macules, often with an oval or “ash-leaf” appearance, commonly associated with tuberous sclerosis (TSC), a condition characterized by benign tumors in multiple organs. While all the clinical features of TSC may not be apparent in the first year of life, the median age of presentation is over 6 months of age, most commonly with seizures, infantile spasms, or several macules. Cardiac rhabdomyoma, often found on prenatal ultrasound (US), is the second most common presentation. More than three macules, at least 5 mm in diameter, are part of the criteria for TSC. Should the clinician suspect TSC in an infant with several criteria, the infant should be referred for molecular genetic testing (Fig. 104.14).
NEONATAL HEAD AND NECK EMERGENCIES
KEY POINTS
 Neonates may develop hemorrhagic shock from intracranial or subgaleal bleeding.
Neonates may develop hemorrhagic shock from intracranial or subgaleal bleeding.
 Neonates are obligate nasal breathers until 6 weeks of life. Any nasal obstruction can cause respiratory distress during this period.
Neonates are obligate nasal breathers until 6 weeks of life. Any nasal obstruction can cause respiratory distress during this period.
RELATED CHAPTERS
Signs and Symptoms
• Respiratory Distress: Chapter 66
Medical, Surgical, and Trauma Emergencies
• ENT Emergencies: Chapter 126
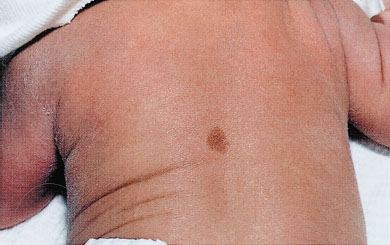
FIGURE 104.13 Café-au-lait macules. (From Fletcher M. Phyical diagnosis in neonatology. Philadelphia, PA: Lippincott-Raven Publishers; 1998.)
Disorders of Head Size, Shape, and Symmetry
CLINICAL PEARLS AND PITFALLS
• Overriding sutures are common in babies born via normal vaginal delivery.
• Neonates can herniate despite an open anterior fontanelle, even without bulging of the anterior fontanelle, if the pathology is confined to the posterior fossa.
• Increase in head circumference accompanied by any sign of increased intracranial pressure (bulging fontanelle and widened sutures) should be investigated.
• Neonates with hydrocephalus generally do not develop papilledema.
• Subgaleal hemorrhage can be an acute life-threatening emergency resulting in acute blood loss and shock. Vacuum extraction and coagulopathy, including vitamin K deficiency, may be predisposing factors.
Current Evidence
Shape, size, and symmetry are factors that must all be considered in the evaluation of the neonate’s head. Macrocephaly, microcephaly, cranial asymmetry, and bulging anterior fontanelle are signs of underlying pathology. Disorders of head shape, size, and symmetry in newborns are often of insidious onset, usually diagnosed prenatally with rare true emergencies.
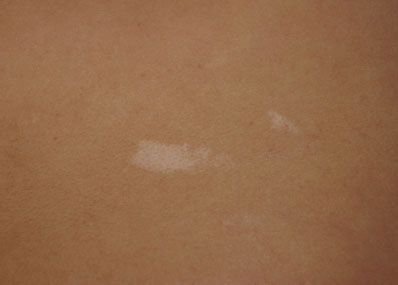
FIGURE 104.14 Ash-leaf macules. (Courtesy of Ilona J. Frieden, MD. In: Chung EK, Atkinson-McEvoy LR, Boom JA, et al., eds. Visual diagnosis and treatment in pediatrics. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010.)
Goals of Treatment
Clinicians need to identify generally innocent etiologies from those that are life-threatening. The primary goals should be early recognition of hemorrhagic shock from intracranial bleeding and increased intracranial pressure. All neonates with disorders of size and shape of head need a thorough physical examination including neurologic examination. Those with significant findings will require head US, computed tomography (CT), or magnetic resonance imaging (MRI). In any patient with significant findings, neonatology, neurology, and/or neurosurgery consultation are warranted. Admission to the intensive care unit is necessary in infants with cardiovascular instability.
Clinical Considerations
Clinical Recognition. The size, shape, and skin discoloration on the head can first be noted by the clinician on inspection of the baby during physical examination. Parents are often unaware of the problem unless the head size is severely enlarged, has prominent skin changes, prominent asymmetry, or the baby is presenting with other symptoms. Vague symptoms may be noted by the parent including poor feeding, irritability, cyanotic episodes (apneic episodes), fever, inability to maintain temperature, or the baby is “just not herself.” Measurement of the head circumference should be routinely performed for infants presenting within the first 2 weeks of life.
Triage Considerations. Neonates with bulging or sunken fontanelle associated with poor feeding, irritability, or temperature instability should be evaluated promptly for meningitis, sepsis, or shock. Associated hypothermia and ill appearance implies neonatal sepsis and demands emergent treatment.
Clinical Assessment. Neonatal disorders of head shape, size, and symmetry are detected by inspection, palpation, and measurement of head circumference. The head should be examined in the midline position, then on each side. Benign dermatologic lesions such as seborrheic dermatitis (cradle cap) (Fig. 104.15), capillary hemangioma, vesicles and ulcers from fetal scalp monitoring electrodes, caput succedaneum (Fig. 104.16), cephalhematoma (Fig. 104.17) and significant conditions such as giant congenital nevi, port-wine stain, and tumors may be immediately identified on the scalp. Figure 104.18 shows a scalp electrode site that has been infected with HSV.
Size. The measured occipitofrontal head circumference should be plotted on the standard CDC full-term growth curve (Link to CDC website for all growth curves: http://www.cdc.gov/growthcharts/clinical_charts.htm). For preterm infants (<37 weeks), the corrected gestational age (CGA = gestational age at birth + postnatal age) should be used to adjust head circumference when using CDC standard curves. Adjustment is continued until 24 to 36 months of chronologic age. Alternatively preterm growth curves such as the Fenton curve may be used (Link to the Fenton Preterm Growth Chart http://www.ucalgary.ca/fenton/2013chart). Head circumference measurement at birth and at initial hospital discharge may vary considerably after 1 week due to subsidence of edema, water loss from the intracranial structures and unmolding of sutures. Accurate measures can be obtain after 1 week and should be followed serially by the pediatrician. Increase in head circumference is normal after 14 days.
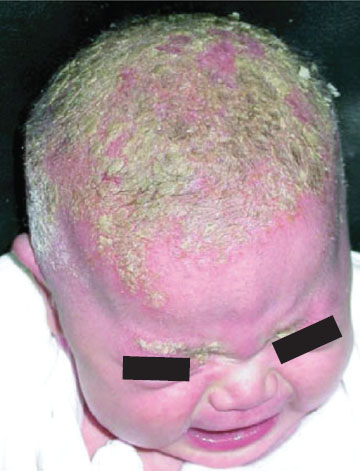
FIGURE 104.15 Severe seborrheic dermatitis (yellow, greasy-appearing plaques). (From Kyle T, Carman S. Essentials of pediatric nursing. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.)
Fontanelles. The anterior fontanelle, situated at the junction of the coronal and sagittal sutures, usually measures about 2 cm × 2 cm (can be up to 5 to 6 cm in its long axis) and normally closes between 9 and 18 months (range 6 to 24 months). The posterior fontanelle, situated at the junction of the lambdoidal and sagittal sutures, generally measures between 0.5 and 1 cm (may be closed at birth in some cases) and usually closes to palpation by 3 to 4 months of age. Enlarged fontanelles may be associated with a variety of conditions, including prematurity, hypothyroidism, rickets, or hydrocephalus. Increased intracranial pressure from infection or hemorrhage produces a full or bulging fontanelle, whereas dehydration produces a depressed fontanelle. A fontanelle that appears full while the infant is supine or crying should be reassessed while the infant is held upright and sleeping or feeding before it is determined to be full or bulging.
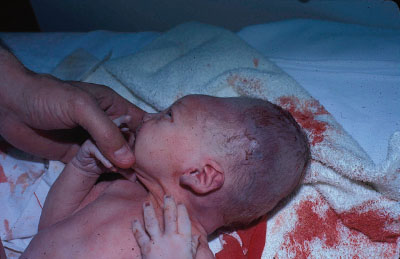
FIGURE 104.16 Caput succedaneum in a newborn infant. Large soft swelling over the vertex, not confined to suture lines. (Courtesy of the late Peter Sol. In: Chung EK, Atkinson-McEvoy LR, Boom JA, Matz PS, eds. Visual diagnosis and treatment in pediatrics. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010.)
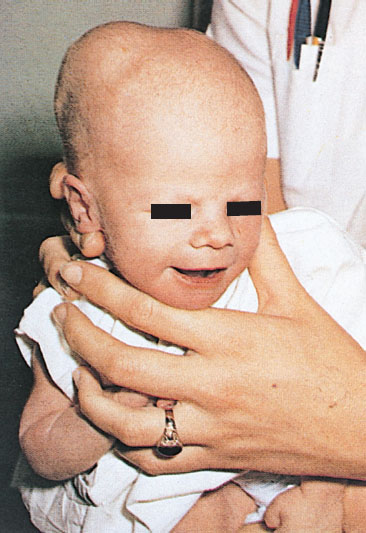
FIGURE 104.17 Unilateral cephalhematoma. Cephalhematoma is a subperiosteal collection of blood and therefore does not cross suture lines. (From O’Doherty N. Atlas of the newborn. Philadelphia, PA: JB Lippincott; 1979:136, 117 and 143.)
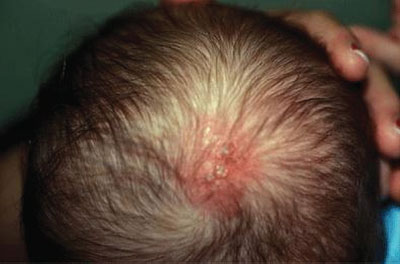
FIGURE 104.18 Neonatal herpes simplex. Herpetic vesicles and erythema at the site of scalp electrode placement. (Courtesy of Shirley P. Klein. In: Chung EK, Atkinson-McEvoy LR, Boom JA, Matz PS, eds. Visual diagnosis and treatment in pediatrics. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010.)
Symmetry. Molding of the skull bones during the vaginal delivery process is a common cause of temporary asymmetry, as is scalp edema or caput succedaneum (Fig. 104.16). Caput succedaneum is an ill-defined, generalized swelling of the soft tissues of the scalp that was the presenting part during a vertex delivery. It is composed of edematous skin containing extravasated serum or blood that extends across suture lines. Presence of localized petechiae and purpura can occur. Generally, both caput succedaneum and skull molding spontaneously resolve by 7 to 10 days of age.
Overriding cranial sutures, or molding, that persists beyond 7 to 10 days may be a sign of underlying brain pathology and deserve further evaluation. Ridging or prominence of cranial sutures may be a sign of craniosynostosis, a premature fusion of cranial sutures. Overriding sutures are ballottable, but if the sutures are rigid and have a heaped-up solid closure, radiographs, or CT scan should be conducted to rule out craniosynostosis. Soft areas, craniotabes, are occasionally found on palpation of the parietal bones during the first several days of life, especially in premature infants. Soft areas noted in the occipital region may be suggestive of osteogenesis imperfecta or other syndromes and should be investigated.
Management. Disorders of head size/shape and symmetry can be diagnosed by a variety of modalities. Skull x-rays should be obtained for assessment of craniosynostosis. Head US is the modality of choice for intracranial hemorrhage in infants less than 6 months of age in whom the anterior fontanelle is patent. CT scan is a good modality for assessment of intracranial and extracranial hemorrhages, skull fractures, and bony lesions, particularly with the advent of newer technology that minimizes exposure to radiation. MRI is preferable for assessment of congenital anomalies of brain, although emergency availability and the need for deep sedation sometimes preclude its routine use. Often neonates can be swaddled and fed for routine MRIs without the need for sedation.
Macrocephaly (Large Head)
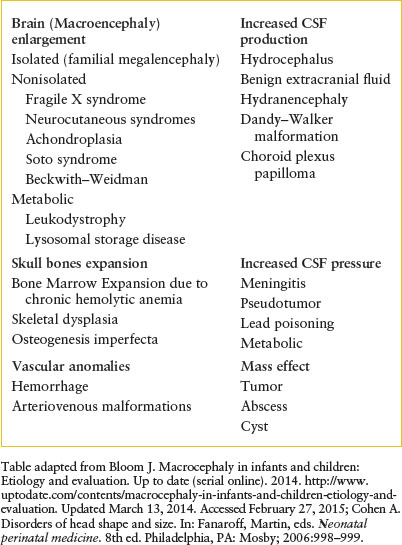
Macrocephaly is defined as an increase in the measured occipitofrontal head circumference greater than the 97th percentile, or greater than two standard deviations above the mean head circumference for age and sex. It is caused by a heterogeneous group of conditions which fall into three categories (Table 104.1): increase in brain volume (megalencephaly) (Fig. 104.19), increase in CSF (either benign extracerebral collections [BECC] or hydrocephalus), or presence of a mass (e.g., brain tumor, abscess, cyst, or subdural collection). Differentiating acute conditions from progressive lesions will depend on evaluation of the perinatal history, family history, progression of the enlargement, delay or loss of developmental milestones, and neurologic signs and symptoms. Symptoms of increased intracranial pressure include poor feeding, vomiting, and irritability. Signs include bulging fontanelle, lethargy, stupor, encephalopathy, cranial nerve palsy, or hypertonia with hyperreflexia. Neurocutaneous lesions may be present for some progressive conditions. Seizures may be subtle. Evaluation for genetic syndromes such as Neurofibromatosis-1, Sturge–Weber Syndrome, TSC, and Fragile X should be performed if the baby appears to have other signs and symptoms. These conditions will require consultation with a neurologist and a geneticist. BECC should be differentiated from subdural collections caused by child abuse. MRI shows that BECCs are in the subarachnoid space, have normal CSF intensity, and are associated with widened cerebral sulci. In contrast, subdural hematomas are of mixed density or more dense than CSF, and compress the adjacent sulci or produce mass effect on the ventricles. MRI provides the best quality for assessment of intracranial pathology; however, US imaging and CT scan (with or without contrast) can also be helpful. The choice between these studies can be determined in conjunction with pediatric neuroradiologist and neurologist depending upon the suspected etiology, acuity of symptoms, need for sedation, and availability. The lack of radiation exposure is a major advantage of MRI over CT. Neurology and neurosurgical consultations are essential to determine further testing. Any newborn with signs of increased intracranial pressure or seizures should have emergent evaluation and inpatient admission.
TABLE 104.1
EXAMPLES OF MACROCEPHALY IN NEONATES AND INFANTS
Hydrocephalus
Hydrocephalus in a newborn (accumulation of intracranial CSF) develops from an imbalance between production and reabsorption of CSF. The majority of cases result either from obstruction to the CSF circulation, called obstructive or noncommunicating hydrocephalus, or from failure to absorb CSF by the arachnoid villi and cisterns, called communicating hydrocephalus. Both of these conditions can be caused by a congenital lesion (e.g., aqueductal stenosis (Fig. 104.20), Dandy–Walker malformation, or Arnold–Chiari II malformation), or by acquired disease (e.g., posthemorrhagic, postmeningitic [bacterial, viral, fungal, or parasitic]), tumor-related compression (least common), or ventriculoperitoneal shunt malfunction. Overproduction by a choroid plexus papilloma or villous hypertrophy is rare. The neonatal course may identify prematurity with intraventricular hemorrhage and posthemorrhagic hydrocephalus, with or without ventriculoperitoneal shunt placement. Occasionally, postnatal enlargement of the head occurs after the baby is at home and may present with rapidly increasing head circumference, full fontanelle, and splayed sutures. Presence of open squamosal suture (between temporal and parietal bones) is a useful sign. Signs of increased intracranial pressure are more common after 2 years of age due to presence of open sutures in neonates. Papilledema is rare in neonates. In unchecked cases, the head enlargement will cause bossing of the forehead, sunset appearance of the eyes (downward deviation of eyes with prominence of the sclera) due to compression of the gaze center in the tectum (Fig. 104.21), esotropia due to paresis of the abducens nerve (VI), thinning of the skull bones leading to a “cracked pot” sound on percussion (Macewen sign). Head US is the most rapid diagnostic tool for evaluation. MRI gives detailed examination, can show meningeal enhancement in case of meningitis and delineate complications (intracranial abscess, venous thrombosis, and empyema). CT scan can be used to provide additional detail, evaluate patients for shunt malfunction and intracranial calcifications in cases with congenital infection. Neurosurgical consultation will be required. Treatment of hydrocephalus should be directed toward ventricular decompression, or resection of the mass, or both. Prognosis is determined by the underlying etiology and timing of treatment. Shunt complications includes occlusion of proximal or distal ends, fracture, displacement, migration, and infection.
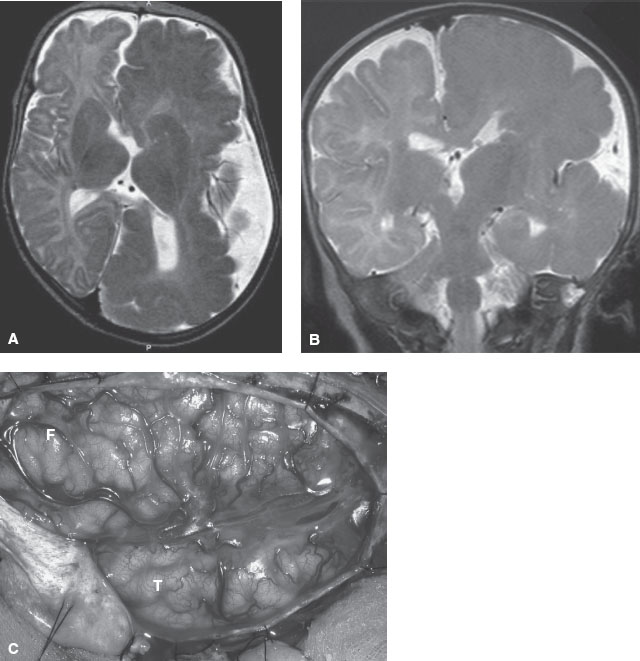
FIGURE 104.19 MRI image showing hemimegalencephaly progression in a 6-month-old child. Axial (A) and coronal (B) T2-weighted magnetic resonance imaging (MRI) images show markedly enlarged left cerebral hemisphere with thickened gyri and smooth surface, consistent with HME. The left lateral ventricle and caudate head are deformed. White matter is diffusely hypointense compared with the unaffected right cerebral hemisphere, suggesting accentuated myelination. C: Intraoperative photograph of the region of HME oriented with the frontal lobe (F) in the top left and the temporal lobe (T) in the lower middle portions of the image. Notice the diffuse cortical disorganization of all gyri. This child had a monozygotic twin who was normal. (Panel A and C: From Engel J, Pedley TA, Aicardi J, et al. Epilepsy. 2nd ed. Philadephia, PA: Lippincott Williams & Wilkins; 2007. Panel B: From Salamon N, Andres M, Chute DJ, et al. Contralateral hemimicrencephaly and clinical-pathologic correlations in children with hemimegalencephaly. Brain 2006;129:352–365; with permission of the editor of Brain and Oxford University Press.)
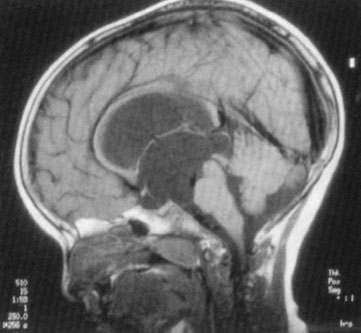
FIGURE 104.20 Congenital aqueductal stenosis and hydrocephalus. Congenital aqueductal stenosis. Midsagittal T1-weighted MR image shows marked dilatation of the lateral and third ventricles with a normal-sized fourth ventricle. (From Eisenberg RL. An atlas of differential diagnosis. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2003. Original: Reprinted with permission from Neuroradiology Companion by M Castillo, Lippincott-Raven, © 1999.)
FIGURE 104.21 Acute hydrocephalus with typical sun-setting appearance of the eyes, increased head circumference, and dilated scalp veins. (From Fleisher GR, Ludwig W, Baskin MN. Atlas of pediatric emergency medicine. Philadelphia, PA: Lippincott Williams & Wilkins; 2004.)
Vein of Galen Malformation
Vein of Galen malformation is a rare massive intracranial arteriovenous shunt caused by failure of the vein of Markowski to involute in utero. Neonates present with a loud bruit over the skull and signs of high output congestive heart failure, cerebral ischemia, and infarction from vascular steal. In infancy, children present with macrocephaly due to hydrocephalus caused by obstruction of CSF flow, seizures, and developmental delay. Doppler US, contrast-enhanced CT, computed tomographic angiography, or MRI angiography may be needed to diagnose the site of intravascular shunting. Treatment is directed at endovascular obliteration and cardiac support if heart failure is present.
Microcephaly (Small Head)
Microcephaly is defined as a decrease in the measured occipitofrontal head circumference below the 3rd percentile or below two standard deviations from the mean head circumference for age and sex. The term microcephaly groups together disorders of different etiologies that are classified as primary microcephaly (caused by early insults to the developing brain that affect neuronal development or migration, usually genetic, chromosomal, or environmental) and secondary microcephaly (caused by a late insult to a normally developed brain, including hypoxia, ischemia, infection, trauma, or metabolic disorders) (Table 104.2). Patients present at a mean age of 7 to 8 months with neurodevelopmental delays (65%) and acute seizure disorder (43%). Seizures may be recurrent and difficult to control. History and physical examination are able to determine causality in only one-third of cases. History of prematurity, perinatal history of maternal infection, congenital infection, drug and alcohol abuse, environmental exposures, family history of metabolic or genetic diseases, and birth data relating to possible anoxic injury may be of help. A thorough neurologic examination is warranted. Management of microcephaly usually requires a multidisciplinary approach. Control of acute seizures and referral to pediatric neurologist is recommended. Specific testing, for example, MRI, audiology, metabolic and genetic testing (karyotype, chromosomal microarray, chromosomal breakage analysis and sequencing) can then be done.
TABLE 104.2
EXAMPLES OF PRIMARY AND SECONDARY MICROCEPHALY

Abnormalities of Head Shape
Craniosynostosis
Craniosynostosis occurs because of premature closure of one or more of the cranial sutures, resulting in an abnormality in the shape of the head perpendicular to the closed suture. It is occasionally accompanied by facial dysmorphism. In cases where multiple sutures are involved, infants may develop increased intracranial pressure and neurocognitive impairment. The incidence of craniosynostosis is approximately 1 per 2,000 live births; sagittal synostosis being the most common (40% to 55%). Primary craniosynostosis occurs when there is primary closure of a suture and may be nonsyndromic (isolated) or syndromic (with associated anomalies as in Apert, Crouzon, and Pfeiffer syndromes). Secondary craniosynostosis is caused by a systemic disorder (e.g., thalassemia). Pathogenesis is thought to be multifactorial but fundamentally related to premature signaling of the dura to the sutures to close prematurely before completion of brain growth. Fibroblastic growth factor receptor (FGFR1 and 2) mutations have been implicated in both the nonsyndromic and syndromic types. Diagnosis of craniosynostosis is made on clinical appearance of the shape (Fig. 104.22). Palpation reveals a ridge along the closed suture and compensatory bossing, or deformity on the contralateral side. In brachycephaly syndromes, the orbit is shallow with proptosis of the eyes, and there is a characteristic “parrot beak” nose (short, stubby, and upturned). Physical examination may reveal polysyndactyly (Apert syndrome) or broad toes and thumbs (Pfeiffer syndrome). Skull radiographs and CT scan are needed to delineate single versus multiple suture involvement. Corrective surgery should ideally occur before 6 months of age. Surgery allows remodeling of the skull, relieves cranial pressure, and improves cosmetic outcome. Prognosis depends on the type of craniosynostosis, the timing of the surgery, and the number of sutures or facial bones involved. All syndromic craniosynostosis usually involve the coronal sutures. Corrective surgeries are directed at expansion of the anterior and posterior cranial vaults and are more complex, requiring a dedicated multidisciplinary craniofacial team.
Deformational Plagiocephaly
Deformational plagiocephaly applies to asymmetry of the cranium caused by flattening of one side of the occiput. Incidence has increased since 1992, after institution of the “back to sleep” campaign. Constant mechanical pressure applied to one side of the head (preferential turning or sleeping on the same side) results in flattening of that side so that the head assumes a parallelogram shape (flattening of the occiput with bossing of the ipsilateral frontal pole). Contributing mechanical forces include in utero forces (bicornuate uterus or twin pregnancy), or postnatal conditions, such as preferential head turning torticollis or vertebral anomalies. At birth, infants have round heads but will develop this shape after a few months. The ipsilateral ear is displaced anteriorly but both ears are in the same horizontal plane. Deformational plagiocephaly should be differentiated from lambdoid suture synostosis, which occurs at birth, with frontal bossing on the contralateral side and posterior and downward displacement of the ears. Skull radiography is usually not necessary if the baby has a typical history and physical examination. Treatment is nonsurgical. Parents are encouraged to alternate head position of the infant and allow infants 30 minutes of tummy time while awake to prevent the occurrence of deformational plagiocephaly. Neck exercises to encourage range of motion have also been found to be helpful in combination with positioning. Alternately, infants with severe deformational plagiocephaly may benefit from helmets, or head bands worn during the day. Corrective surgery is rarely needed for those who do not respond to nonsurgical measures. Prognosis is good and the shape improves after infants gain head control and begin to sit.
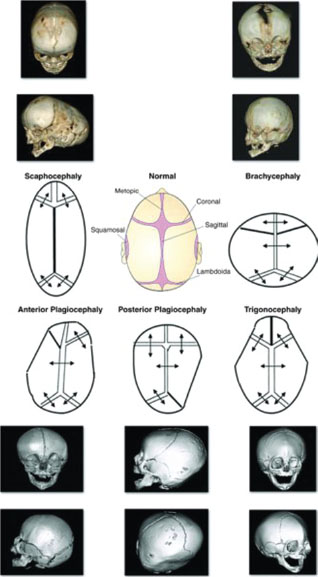
FIGURE 104.22 Morphologic types of craniosynostosis. Diagrammatic views of different morphologic types of craniosynostosis are shown in the center of the figure. The premature closed suture is marked as a bold line. The sutures involved determine the shape of the skull. The diagrammatic view of each synostosis type is matched with the corresponding radiologic findings. The fused suture is seen as a hypertrophic keel or ridge on the x-rays. (From Paul Mongan P, Soriano III SG, Sloan TB, et al. Practical approach to neuroanesthesia. Philadelphia, PA: Lippincott Williams & Wilkins; 2013.)
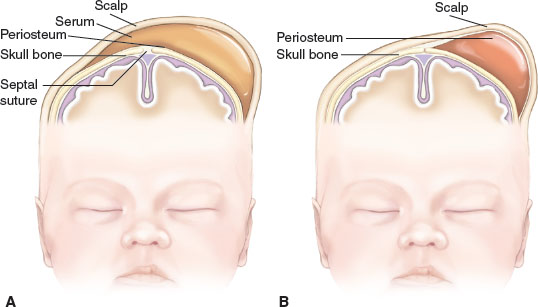
FIGURE 104.23 A: Caput succedaneum involves the collection of serous fluid and often crosses the suture line. B: Cephalhematoma involves the collection of blood and does not cross the suture line. (From Ricci SS. Essentials of maternity, newborn, and women’s health nursing. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.)
Perinatal Birth Injuries to Head and Neck
Goals of Treatment
Trauma during labor and delivery can be related to maternal, fetal, and obstetric factors. Traumatic deliveries occur as a result of prolonged labor, large fetus, prematurity, and abnormal presentation, and as a result of assisted deliveries, for example, vacuum extraction and forceps delivery. Although the majority of these injuries are diagnosed prior to discharge from the newborn nursery and are self-limiting, some are serious and potentially lethal. The primary goals of management should be to recognize benign injuries and differentiate them from those caused by nonaccidental trauma and manage any acute crises, for example, acute hemorrhagic shock.
CLINICAL PEARLS AND PITFALLS
• Cephalhematomas are subperiosteal hemorrhages that do not cross the suture lines. They should not be routinely incised or aspirated.
• Subgaleal hemorrhage can lead to hypovolemic shock and can present days after birth.
• Traumatic facial nerve palsy is unilateral and will usually recover spontaneously.
• Neck masses in a neonate need evaluation for airway proximity and compression.
Cephalhematoma
Cephalhematoma (Fig. 104.17) is a subperiosteal hemorrhage occurring commonly over a parietal bone, distinguished from a caput succedaneum by the fact that the swelling never crosses suture lines (Fig. 104.23). It occurs in 0.4% to 2.5% of live births due to rupture of blood vessels traversing the skull to the periosteum. The overlying skin is intact with no petechiae or hemorrhage. Cephalhematomas often feel fluctuant and maybe bordered by elevated ridges of surrounding tissue giving a false sensation of a skull depression. They may be associated with intracranial hemorrhage and 5.4% are also associated with linear skull fractures.
Cephalhematomas often become prominent after the immediate newborn period when scalp edema subsides. Most commonly, a cephalhematoma is unilateral, but it can be bilateral. They resolve slowly over 4 to 6 weeks, possibly with calcification and the formation of a hard bump on the scalp that may be a source of great concern to parents. Occasional complications resulting from the breakdown and resorption of large hematomas are hyperbilirubinemia or anemia. No therapy is required for uncomplicated lesions. Routine incision or aspiration of a cephalhematoma is contraindicated due to the high risk of introducing infection. Rare complications of localized infection of cephalhematoma include osteomyelitis, meningitis, and venous sinus thrombosis.
Subgaleal Hemorrhage
Subgaleal hemorrhage (SGH) refers to hemorrhage into the soft tissue space between the galea aponeurotica and the periosteum (Fig. 104.24). Rupture of emissary veins within this extensive space results in hemorrhage across the whole cranial vault from the orbital ridges to the nape of the neck. This space can hold 260 mL of blood (which could exceed the entire blood volume of a full-term baby) resulting in hemorrhagic shock. Newborns with SGH have a history of difficult instrumental delivery using vacuum suction or forceps (64% to 91%). Other predisposing factors include coagulopathies (hemophilia and Christmas disease), vitamin K deficiency, macrosomia, and dystocia.
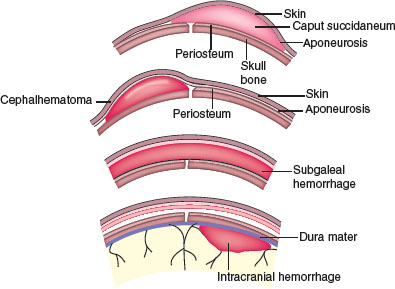
FIGURE 104.24 Subgaleal hemorrhage and normal caput, cephalhematoma, and intracranial hemorrhage. (From Gibbs RS, Karlan BY, Haney AF, et al. Danforth’s obstetrics and gynecology. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.)
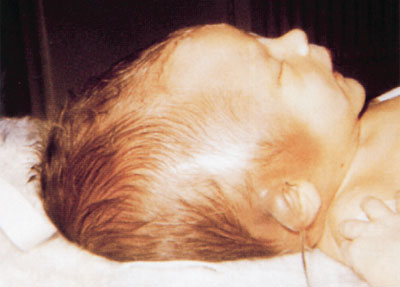
FIGURE 104.25 Subgaleal hematoma. Discoloration and swelling extends across suture lines onto the neck, even onto the ear, causing protuberance of the pinna. (From Fletcher MA. Physical diagnosis in Neonatology. Philadelphia, PA: Lippincott–Raven Publishers; 1998:185.)
Subgaleal hemorrhage develops insidiously and may not be apparent until several hours or even days after delivery and until blood loss is extensive. Discoloration of the scalp occurs very late as blood collects deep beneath the aponeurotic layer. Pallor and weakness may be the only early symptoms of SGH and may be accompanied by a rising pulse rate and increasing respiratory rate. Diffuse pitting swelling of the scalp or a fluctuating mass extending from the occiput posteriorly to ecchymotic orbits anteriorly and displacing the ears is suggestive (Fig. 104.25). Hypoperfusion and falling hematocrit in a child who has undergone a difficult extraction should alert the clinician to the possibility of SGH even in the absence of a fluctuant mass. CT scan will demonstrate presence of blood in subgaleal space and rule out associated fractures or intracranial hemorrhages (Fig. 104.26). Patients with SGH require emergency-packed RBC transfusion and may require fresh frozen plasma if PTT is prolonged. Concomitant intravenous administration of vitamin K can be performed. Surgical drainage can be considered as a last resort. SGH with shock has a high mortality rate of 22% to 25%.
Facial Nerve Palsy
Neonatal facial nerve palsy arises either from a traumatic birth injury or from hereditary agenesis of the facial nerve nucleus (Moebius syndrome). Traumatic peripheral nerve injury takes place when the facial nerve is compressed as it exits the stylomastoid foramen or branches within the ramus of the mandible. Compression of the facial nerve may occur during pregnancy or during delivery and may occur as a result of oblique midforceps application, or pressure on the face by extreme prolonged pressure by the sacrum, other fetal parts or uterine fibroid tumors. Peripheral injury is unilateral and may be associated with brachial nerve palsy. Traumatic central nerve injury results from destruction of contralateral brain tissue within the posterior fossa or the temporal bone. Traumatic central injury is rare and can affect other cranial nerves.
Neonates with peripheral facial nerve injury will display injury to both the upper and lower face on the ipsilateral side within the first few days. Typically the forehead is smooth with inability to close the eye, and there is a smooth nasolabial fold and drooping at the corner of the mouth on the paralyzed ipsilateral side. When the baby cries the contralateral corner of the mouth will move. Since other cranial nerves are intact the baby will have no trouble feeding. Peripheral facial nerve palsy starts to recover within days, with complete resolution within weeks to months. Initial supportive care includes corneal protection with petroleum ointment, and eye pad during periods of sleep, or 1% methyl cellulose eye drops every 4 hours. Neonates should be further referred for neurologic evaluation and electrodiagnostic testing to ensure recovery is monitored appropriately. Infants who show poor recovery at 1 year of age may be candidates for surgical intervention.
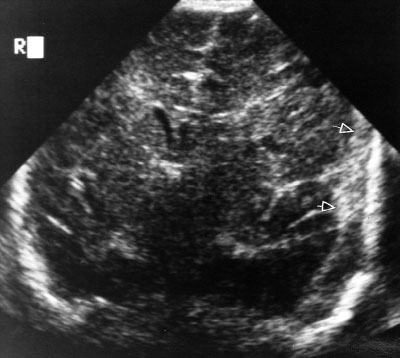
FIGURE 104.26 Noncontrast CT scan showing left-sided subgaleal and subdural hemorrhages in a newborn. Axial noncontrast CT images show subdural hemorrhage along the left leaf of the tentorium (large white arrow), in the interhemispheric fissure near the falcotentorial junction (small white arrow), along the left cerebral convexity subdural space, and in the subcutaneous and subgaleal spaces. Note the severity of the cerebral cortical involvement. (From Barkovich J, Raybaud C. Pediatric Neuroimaging. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.)
Moebius Syndrome
Moebius syndrome is a hereditary absence of the VI and VII nerve nuclei. This results in loss of use of facial muscles and lateral eye motion. Infants are unable to move facial muscles so that the face remains expressionless and immobile. The condition is associated with micrognathia, microstomia, muscle weakness, recurrent aspiration, earlobe deformities, and thoracic and limb deformities. There may be notable dryness of the eyes due to lack of inability to close the eyelids. Neonates with this condition may have difficulty feeding and require respiratory support due to aspiration. Therapy consists of ongoing supportive care. Many newborns are unable to breast-feed or feed from a regular nipple. Infants may require feeding tubes or a Haberman nipple to obtain adequate nutritional intake.
Facial Asymmetry
Facial asymmetry occurs in a newborn because of abnormal in utero position. Commonly, when the face and neck are pressed against the shoulder, a characteristic flattening of the face and angle of the jaw is noted on that side because of displacement of the mandible. This facial asymmetry will resolve spontaneously in a few weeks.
Congenital Torticollis
Congenital muscular torticollis is a positional abnormality of the neck, resulting in abnormal tilting and rotation of the head. It is believed to be secondary to intrauterine positioning or trauma to the soft tissues of the neck during delivery, with resulting ischemia of the sternocleidomastoid muscle secondary to venous occlusion. This leads to edema and degeneration of the muscle fibers with eventual fibrosis of the muscle body. Although congenital muscular torticollis may be noted at birth, it usually manifests at 1 to 2 weeks of age. The incidence is increased in breech presentations and difficult deliveries. Unilateral contracture and fibrosis of the sternocleidomastoid muscle results in a mass and a characteristic head tilt toward the affected side and the chin pointing toward the opposite side (Fig. 104.27). This position is fixed and does not correct with passive attempts to move the head. On examination, a firm, nontender mass may be felt within the body of the sternocleidomastoid muscle which then enlarges within the following 1 to 2 weeks. The baby may develop plagiocephaly in the long term if left untreated. Occipitocervical spine anomalies, such as the Klippel–Feil syndrome (congenital fusion of two or more cervical vertebrae; clinical triad of short neck, limited neck motion, and low occipital hairline), are rare causes of torticollis that present in the newborn period. Spinal and neck x-ray may help in differentiating between these cervical spine anomalies and congenital torticollis. Similarly, CT scan of the neck or soft tissue MRI will help differentiate other soft tissue masses that may have a similar appearance (e.g., hemangioma). Treatment consists of passive stretching exercises of the neck and repositioning toys and mobiles in the crib to stimulate the infant to look toward the side opposite the preferred gaze. Positioning and referral for physiotherapy is recommended as early as possible. Surgery may be considered if there is no response to conservative management. Prognosis improves if the baby is treated before 1 year of age.
FIGURE 104.27 Congenital torticollis. A fibrotic mass located in the sternocleidomastoid muscle of a 1-month-old infant whose mother was concerned about the infant’s head tilt to one side. (From Courtesy of Ellen Deutsch. In: Chung EK, Atkinson-McEvoy LR, Boom JA, Matz PS, eds. Visual diagnosis and treatment in pediatrics. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010.)
Neonatal Eye Disorders
Goals of Treatment
Neonatal eye disorders are infrequent. They, however, do have the potential of causing serious harm if missed. The most challenging part of the eye examination is actually opening the eye of a newborn since any attempt at forcing them open usually meets with marked resistance and blepharospasm. Darkening the examination room, holding the baby upright and gently swaying him or her from side to side or up and down often induces the eyes to open spontaneously. Neonatal ophthalmologic concerns include leukocoria, neonatal conjunctivitis, excessive tearing, scleral and subconjunctival hemorrhages, and uncoordinated eye movements. Absence of red reflex or presence of leukocoria could indicate the presence of retinoblastoma. Cataracts that are not treated may affect visual acuity or lead to visual loss. Physicians need to focus on diagnosis of subtle findings early in their course. Once issues such as absence of red reflex are identified prompt ophthalmology referral is needed.
CLINICAL PEARLS AND PITFALLS
• Retinal hemorrhages are common after labor.
• White or absent red reflex is highly suspicious for retinoblastoma. Patients should be referred to an ophthalmologist.
• The lacrimal duct system is not fully functional until 3 weeks of age so dacryostenosis will often appear at that age.
• Neonates do not focus at birth. Infants start to fixate at 5 to 6 weeks of age.
Scleral and Subconjunctival Hemorrhage
Scleral and subconjunctival hemorrhage are often noted in the newborn as a result of normal delivery or birth trauma. Spontaneous resolution within 1 to 2 weeks is the rule. When the funduscopic examination is performed, similar hemorrhages may be noted on the retina in about 25% of newborns. The presence of retinal hemorrhages should also raise the possibility of intentional trauma. Specifically, the shaken baby syndrome has been associated with flame-shaped retinal hemorrhages and subdural hematomas (see Chapters 95 Child Abuse/Assault and 130 Neurosurgical Emergencies).
Leukocoria
Leukocoria is defined as the presence of a white pupil (Fig. 104.28). A pupillary light reflex is a simple test that should be performed on all newborns. In the normal newborn, a “red reflex” is seen when the ophthalmoscope is held 2 to 3 ft in front of the eyes. A white pupillary light reflex, or leukocoria, occurs when light reflects off a white surface in the eye. It is never normal. An absent or black red reflex is also abnormal. Both indicate presence of an opacity obstructing the light reflection through the layers of the ocular media. Irregular opacities may also produce an irregular reflex. Pupillary examination maybe difficult if the pupils are small or the neonate will not maintain eyes open. In neonates darkening the room and holding up the baby at a 45-degree angle with gentle patting on diaper will help open the baby’s eyes. Occasionally placement of one drop of 0.2% cyclopentolate and 1% phenylephrine (Cyclomydril) dilating eye drops may be of help. Care should be taken with infants who have a history of prematurity or chronic lung disease when using these drops since some infants may develop cardiovascular instability. Leukocoria may be a sign of several conditions of variable severity and prognosis. Causes of leukocoria are listed (Table 104.3). Definite evaluation by slit lamp examination is necessary to determine etiology. CT may be required for some infants to delineate orbital structures, shape of the globe, and other cranial anomalies. Therefore, all infants with an abnormal pupillary light reflex should be referred to an ophthalmologist for a prompt evaluation. Treatment depends on the underlying etiology.
FIGURE 104.28 Unilateral leukocoria in a newborn with congenital rubella. This child has congenital rubella syndrome. Although full term, the baby is small for his gestational age. There is leukocoria due to a cataract in the left eye. (From Garg S. Color Atlas and Synopsis of Clinical Ophthalmology—Wills Eye Institute—Uveitis. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.)
Retinoblastoma
Retinoblastoma is the most common malignant intraocular tumor in neonates, occurring in 1 in 20,000 infants. It can be unilateral or bilateral and can be inherited or sporadic in nature. Retinoblastoma usually presents by 13 to 18 months if not captured earlier on routine examination (50% of cases are diagnosed by finding an abnormality of red reflex during routine examination or by a parent). Mutation or inactivation of the tumor suppressor gene is thought to contribute to its development. Children of affected parents with the inherited or bilateral form have a 45% chance of developing this tumor. As the tumor progresses anteriorly, strabismus due to deterioration of vision occurs. Fundal examination reveals posterior chamber mass with calcification. The globe starts to enlarge because of the large mass or secondary glaucoma. Secondary glaucoma also causes photophobia. Other manifestations include pseudohypopyon and spontaneous intraocular bleeding. CT scan or MRI is necessary for determining the degree of extraocular spread and confirming the diagnosis. Early detection and treatment is of utmost importance because 5-year survival is approximately 98% if the tumor is confined to the globe.
TABLE 104.3
CAUSES OF LEUKOCORIA IN A NEWBORN

Lacrimal Duct Stenosis (Dacryostenosis)
Congenital obstruction of the nasolacrimal duct, dacryostenosis, is the most common cause of excessive tearing in the newborn, occurring in 7% of neonates. The lacrimal duct system is not fully mature and functional until 3 weeks of age so excessive tearing (epiphora) usually appears at that time. Dacryostenosis should be differentiated from congenital or infantile glaucoma, a serious but fortunately rare cause of excessive tearing. Most cases of infantile glaucoma presenting during the first 3 months of life are bilateral, whereas dacryostenosis is usually unilateral. Other causes of epiphora include dacryocystitis, obstruction, congenital absence of puncta, corneal abrasions, and conjunctivitis.
Increased wetness of the affected eye relative to the normal eye, excessive tearing, mucoid eye discharge, and crusting along the eyelid margins are the usual presenting symptoms. Gentle pressure along the medial canthal region over the lacrimal sac may produce a reflux of tears or purulent material onto the surface of the eye, confirming the diagnosis (Fig. 104.29). Infants with glaucoma, in addition to excessive tearing, also present with enlarged cornea, rhinorrhea, photophobia, blepharospasm, and corneal haziness. The cornea may be inspected after instillation of fluorescein dye to rule out a corneal abrasion as the reason for the excessive tearing.
Uncomplicated cases of nasolacrimal duct obstruction should be managed with gentle cleansing of the eyes, followed by local massage of the nasolacrimal duct, several times per day, to prevent stagnation of tears in the lacrimal sac. Topical ophthalmologic antibiotic ointments should be prescribed if there is associated conjunctivitis, purulent discharge, or dacryocystitis. Systemic antibiotics occasionally may be needed for cases with cellulitis. Ninety percent of cases resolve within the first year. Referral to an ophthalmologist for probing should be considered for children with a prolonged course. Suspected cases of infantile glaucoma require immediate ophthalmologic evaluation under anesthesia including tonometry. Neonates with congenital glaucoma are treated with surgery.
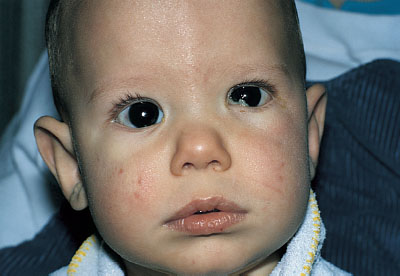
FIGURE 104.29 Congenital dacryostenosis. The child’s parents complained of epiphora of both eyes since the child’s birth. The right eye cleared spontaneously, but the left eye continued to tear, with morning crusting and heavy matting. (From Tasman W, Jaeger E. The Wills Eye Hospital Atlas of Clinical Ophthalmology. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001.)
Neonatal Conjunctivitis (Ophthalmia Neonatorum)
The major causes of neonatal conjunctivitis, or ophthalmia neonatorum, are chemicals, chlamydia, bacteria, and viruses. Chlamydial conjunctivitis is the most common conjunctivitis in neonates. The time of onset of symptoms after birth can help identify the causative agent. Mild nonpurulent inflammation of the conjunctivae that begins 12 to 24 hours after birth is typically caused by the prophylactic 1% silver nitrate instilled at birth. This chemical conjunctivitis usually resolves by 48 hours of age. The incidence of chemical conjunctivitis has decreased since erythromycin eye drops (0.5%) have replaced silver nitrate drops in the United States and Canada. Chemical conjunctivitis may still be seen in developing countries where silver nitrate is used.
Neisseria gonorrhoeae conjunctivitis generally appears 2 to 5 days after birth, whereas conjunctivitis caused by Chlamydia trachomatis presents between 5 and 14 days after birth because of its longer incubation period. Gonococcal infection may be delayed beyond 5 days of age because of partial suppression by the prophylactic drops instilled at birth. Gonococcal infection usually manifests as marked inflammation of the eyelids, chemosis, and copious purulent discharge. Presentation of chlamydial infection, which is primarily localized to the palpebral conjunctiva, can vary from mild inflammation to severe swelling of the eyelids with copious discharge. Of neonates with chlamydial conjunctivitis, 10% to 20% have chlamydial pneumonia, which can either occur simultaneously with the eye infection or up to 4 to 6 weeks later. Gonococcal conjunctivitis is considered a medical emergency because the infection can spread to the cornea, producing corneal ulceration and perforation. HSV is a less common cause of neonatal conjunctivitis. The presence of skin lesions can help in the diagnosis. Gram stain and cultures are essential in the evaluation of neonatal conjunctivitis. Giemsa staining from conjunctival scrapings and direct immunofluorescence antibody test can identify chlamydial disease. Gonococcal conjunctivitis should be treated immediately with intravenous penicillin or a third-generation cephalosporin in addition to topical antibiotics and irrigation. Chlamydial conjunctivitis should be treated with oral azithromycin. Topical drops maybe helpful in clearing debris but are not effective (see also Chapter 131 Ophthalmic Emergencies).
Transient Neonatal Strabismus
Intermittent esotropia or exotropia may be noted in normal infants during the first 2 to 3 months of life. Esotropia (crossed eyes) is more common than exotropia. Deviations secondary to neuromuscular immaturity generally resolve spontaneously by 3 to 4 months of age. Eye deviations that are constant rather than intermittent, or that do not resolve by 3 months of age, warrant referral for a full ophthalmologic examination (including fundus examination) to rule out pathology. Esotropia can occur when visual acuity is impaired (e.g., corneal abrasions, cataracts, chorioretinitis, and tumors), or due to paralysis of the lateral rectus muscle. In these cases, early treatment of strabismus improves the prognosis for cosmetic appearance and will prevent amblyopia.
In many infants, a broad, flat nasal bridge and prominent epicanthal folds may obscure a medial portion of the sclera near the nose and create the appearance of esotropia. This pseudostrabismus, or apparent deviation of the eyes, is an illusion that can be dispelled by the finding of symmetric corneal light reflexes.
Disorders of the Neonatal Nose and Mouth
Goals of Treatment
Many neonatal nose and mouth disorder can be recognized immediately after birth or early in their nursery stay. Infants occasionally develop symptoms of acute airway obstruction and respiratory distress after discharge home. Airway obstruction can occur at any level from nose to trachea, leading to variable degrees of distress. Physicians must determine how much the obstruction is affecting the baby and if emergency airway management is needed.
CLINICAL PEARLS AND PITFALLS
• Neonates are obligate nasal breathers until 6 weeks of life. Any nasal obstruction can cause respiratory distress during this period.
• The ability to successfully pass a nasogastric tube bilaterally rules out choanal atresia.
• Natal teeth can be an aspiration risk. If loose, they should be extracted.
• Cysts or masses in the neck should be immediately assessed for acute airway obstruction.
Nasal Obstruction
Neonates are obligate nasal breathers up to 4 to 6 weeks of age. Any nasal obstruction causing bilateral mucosal swelling will cause significant symptoms of obstruction and respiratory distress. This is commonly seen in infants with upper respiratory tract infections. Simple nasal obstruction should be differentiated from unilateral and bilateral choanal atresia. Bilateral choanal atresia occurs in 1 in 7,000 live births. Associated anomalies such as CHARGE syndrome (Coloboma, Heart disease, choanal Atresia, Retarded development, Genital hypoplasia, Ear anomalies with hearing loss) occur in 20% to 50% of neonates with bilateral choanal atresia. Unilateral atresia is more common than bilateral involvement. Symptoms of bilateral choanal atresia vary from mild cyanosis during feeds to severe hypoxia and respiratory distress. The onset is shortly after birth. Cyanosis occurs when the mouth is completely occluded during feeds and improves with crying. Inability to pass a 6F catheter bilaterally through the nares is suspicious. Smaller soft catheters may coil in the nares and give a false impression of patency. Visualizing or palpating the nasogastric tube in the mouth will also confirm passage and patency. Clinicians should also avoid unnecessary mucosal swelling by repeated forceful trials since this will also make it difficult to rule out obstruction. Unilateral choanal atresia presents with intermittent nasal obstruction which may pass unnoticed at birth. Bedside endoscopic nasal examination can be performed by trained personnel or otolaryngologist. A CT or MRI scan will confirm presence of choanal atresia, differentiate unilateral from bilateral, differentiate between choanal stenosis and atresia, and determine whether the lesion is boney or membranous (Fig. 104.30). MRI minimizes radiation exposure and can determine presence of nasal masses. Simple nasal obstruction due to edema is managed conservatively with humidification, saline drops, and gentle suctioning. Vigorous suction can worsen mucosal edema. Intranasal steroid drops should be used with caution as prolonged use may lead to paradoxical worsening. Neonates with bilateral choanal atresia should have an oral airway placed to maintain mouth patency and relieve the obstruction. Endotracheal intubation is warranted if there is severe respiratory distress or if there is continued obstruction due to failure to maintain patency of the airway. Otorhinolaryngology consultation and hospital admission are necessary. A genetic workup is warranted for diagnosis of associated conditions.
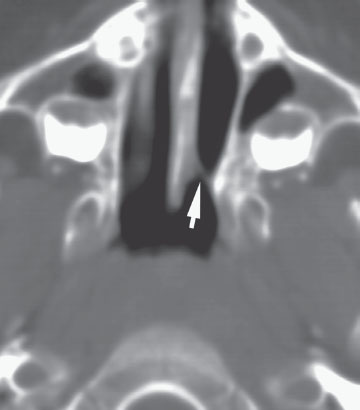
FIGURE 104.30 Choanal atresia in a newborn with difficulty breathing. Axial CT scan shows a widely patent right nasal passage. The left nasal passage is occluded by a thin, ossified septum (arrow). (From Siegel MJ, Coley B. Core Curriculum: Pediatric Imaging. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.)
Normal Neonatal Mouth Findings
Common normal findings in the oropharynx include natal teeth and benign gingival cysts. The incidence of natal teeth (teeth present at birth) is about 1 in every 3,000 live births. The mandibular central incisors are the most commonly affected teeth. One single tooth usually erupts; eruption of two is rare (Fig. 104.31). Because most natal teeth are primary teeth that have erupted early, they should be extracted only if they are loose and pose a danger of aspiration, cause discomfort to the mother or child during nursing, or are confirmed to be supernumerary by focused radiographic examination. Consultation with a pediatric dentist is strongly recommended.
Benign gingival cysts are found in 75% of newborns. Epstein pearls are usually single, small, white, keratin-filled cysts found along the midline of the palate (Fig. 104.32). Bohn nodules are mucous gland cysts that appear as multiple, firm, grayish white lesions along the gums and occasionally on the palate (Fig. 104.33). Dental lamina cysts are formed by remnants of dental lamina epithelium and appear as small, cystic lesions along the crests of the mandibular and maxillary mucosa. They are usually larger and more lucent than both Epstein pearls and Bohn nodules. These cysts generally disappear by 4 weeks of age.
Cleft Lip and Palate
Defects in the formation of the lip (cleft lip) are obvious on even casual visualization. They occur in 0.8% of live born infants. Cleft lip is usually lateral in position and maybe isolated or accompanied by a cleft palate. Isolated cleft palate may be more difficult to detect without focused inspection of the mouth, but should be considered in any neonate with feeding difficulties. Median cleft lip and palate is suspicious for other associated midline defects such as holoprosencephaly. Cleft lip and palate can occur in many diverse syndromes. Physical examination will delineate the extent of the defect and should also determine presence of associated anomalies. Presence of pits in the lower lip is suggestive of Van Der Woude syndrome (an autosomal recessive disorder). These infants should be referred for eventual surgical repair and for monitoring of feeding techniques. Genetics referral is also recommended if other accompanying features are suggestive of other syndromes.
FIGURE 104.31 Natal teeth. Bilateral lower central incisors in a neonate. (Courtesy of Denise A, Salerno, MD, FAAP. In: Chung EK, Atkinson-McEvoy LR, Boom JA, Matz PS, eds. Visual diagnosis and treatment in pediatrics. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010.)
Neck Cysts
Congenital neck lesions may present during infancy or sometimes later in childhood. The most common lesions include thyroglossal duct cysts (midline in the neck and inferior to the hyoid bone), dermoid cysts (midline tongue lesions), branchial cleft cysts (along the lateral neck), and cystic hygromas (usually located behind the sternocleidomastoid muscle in the supraclavicular fossa; two-thirds of cystic hygromas are present at birth). Obstruction of the airway may occur as a result of mass effect when cysts increase in size. This can be insidious in onset, occurring months after birth. Infection in any of these cysts can cause rapid increase in size. Differential diagnosis of neck masses in a neonate includes reactive lymphadenitis, thyroid cysts, lymphatic or vascular malformations, and tumors. Ultrasonography (US) is a rapid tool for evaluation of neck cysts in an afebrile child. US will confirm presence of a thyroglossal cyst and can also rule out ectopic thyroid tissue and visualize a normal thyroid. CT scan with contrast gives a better delineation of extent of cystic lesions such as cystic hygroma but should be used judiciously because of the high dose of radiation. MRI is helpful in determining extent of vascular lesions. Assessment of airway for acute obstruction is of utmost important. Surgical excision or sclerotherapy is the treatment of choice depending on the type and impact of the lesion.
FIGURE 104.32 Benign gingival cysts. Epstein pearls. (From O’Doherty N. Atlas of the Newborn. Philadelphia, PA: JB Lippincott; 1979.)
FIGURE 104.33 Bohn nodule. Pearly nodule on the gum of a neonate. (From Fletcher MA. Physical diagnosis in neonatology. Philadelphia, PA: Lippincott–Raven Publishers; 1998:216.)
NEONATAL CARDIAC EMERGENCIES
This section focuses on ductal-related structural heart disease in the neonate presenting in the first few weeks of life. Please see Chapter 94 Cardiac Emergencies for emergency management of arrhythmias, shock, and acquired heart disease.
KEY POINTS
 Congenital heart lesions can present as late as 2 to 3 weeks of age as the ductus arteriosus closes.
Congenital heart lesions can present as late as 2 to 3 weeks of age as the ductus arteriosus closes.
 Tachypnea may be the only sign of congestive heart failure.
Tachypnea may be the only sign of congestive heart failure.
 Femoral pulses may be palpable with aortic coarctation because of collateral vessels. Instead palpate distal lower extremity pulses and obtain 4-extremity blood pressures.
Femoral pulses may be palpable with aortic coarctation because of collateral vessels. Instead palpate distal lower extremity pulses and obtain 4-extremity blood pressures.
 Prostaglandin infusion may be lifesaving even if the specific cardiac diagnosis is not known.
Prostaglandin infusion may be lifesaving even if the specific cardiac diagnosis is not known.
 Right-to-left shunts often are not associated with heart murmurs.
Right-to-left shunts often are not associated with heart murmurs.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


