INTRODUCTION
Metabolic emergencies are challenging childhood disorders, often presenting with nonspecific signs and symptoms that may mimic more common conditions such as sepsis. Delay in accurate diagnoses can lead to significant morbidity and mortality, whereas early aggressive management based on probable diagnosis can be lifesaving and reduce long-term neurologic sequelae.
In any healthy neonate, sudden acute deterioration should prompt consideration of metabolic disease. Vomiting, altered mental status, and poor feeding are the most common clinical features of metabolic emergencies. Appropriate initial management can be started in the ED without definitive diagnosis. This chapter reviews the most common metabolic disorders presenting as acute decompensation in the young infant and the ED treatment. Hypoglycemia is discussed separately. Congenital adrenal insufficiency is included here because of the overlap in presentation with other inherited metabolic disorders and the importance of prompt recognition and treatment in the critically ill neonate. Inherited metabolic disorders that present in later childhood, such as lysosomal storage disease, are often diagnosed and managed outside of the ED, and these disorders are not included here.
HYPOGLYCEMIA
Hypoglycemia is a plasma glucose level of <45 milligrams/dL (2.6 mmol/L) in any symptomatic child or <35 milligrams/dL (<1.9 mmol/L) in an asymptomatic neonate.1,2 Intellectual performance is poor at 18 months in premature infants with persistent serum glucose <47 milligrams/dL,3 and MRI of infants with episodes of hypoglycemia demonstrates patterns of brain injury.4,5,6 This has led to the recommended treatment threshold of 45 milligrams/dL (2.6 mmol/L) for neonates, who may be at higher risk of poor neurologic outcomes than older infants and children.7 Hypoglycemia in children requiring resuscitation is associated with increased mortality, and hypoglycemia in the setting of seizures is associated with poor neurologic outcomes.2,8
Neonates are born with 60% to 80% of maternal glucose levels. Within 2 to 4 hours, neonates begin to regulate their own serum glucose. Maintenance of serum glucose depends on intake and endogenous gluconeogenesis and glycogenolysis mediated by various hormones. Serum glucose level is affected when there is an imbalance between insulin (hypoglycemic hormone) and its counterregulatory hormones cortisol, growth hormone, glucagon, and epinephrine (hyperglycemic hormones). Insulin stimulates cellular glucose uptake and suppresses lipolysis, whereas hyperglycemic hormones stimulate lipolysis and glycogenolysis. Excess insulin (hyperinsulinemia) results in hypoglycemia with absence of urinary ketones. Hypoglycemia in the neonate or infant may result from inadequate oral intake, excess insulin, deficient hyperglycemic hormones (e.g., growth hormone or adrenal hormone deficiency), disorders of fatty acid oxidation or carbohydrate metabolism, aminoacidopathies and organic acidurias (due to inhibition of gluconeogenesis), or systemic infection (sepsis). Infants of diabetic mothers, postterm infants, and large for gestational age infants are at risk for hypoglycemia due to excess fetal insulin levels in response to elevated maternal serum glucose levels, whereas premature infants or those small for gestational age are at risk due to inadequate glycogen stores.9,10,11
Neonates and infants with hypoglycemia typically present with alterations in mental status. Nonspecific symptoms include poor feeding, an abnormal or high-pitched cry, cyanosis, and hypothermia, and varying degrees of irritability and jitteriness or lethargy.12 Severe hypoglycemia may result in coma or seizures.
Inquire about maternal complications during pregnancy (including gestational diabetes, growth retardation, and infections) as well as any history of prior spontaneous abortions or early infant deaths, which may signal inherited metabolic disease. Obtain a detailed feeding history, and document duration and progression of symptoms as well as the presence of associated signs and symptoms of vomiting, diarrhea, abnormal urine output, jaundice, and temperature instability.
The classic signs of hypoglycemia seen in older children and adults are a result of the hyperglycemic hormones and include signs of adrenergic stimulation such as tachycardia, diaphoresis, tremor, anxiety, and tachypnea. Neonates and infants may not manifest these signs, and lethargy, apnea, or seizures may be the prominent finding. A careful neurologic examination should focus on mental status, tone, and reflexes, and may reveal focal neurologic deficits similar to Todd’s paralysis in cases of prolonged, severe hypoglycemia. Seizures may be noted.
A complete physical examination is important to search for primary and secondary causes of hypoglycemia. Document weight and compare with birth weight (if known) in the neonate. Macrosomia or growth retardation may provide a quick clue to potential cause. Dysmorphic features should be documented. Fever or hypothermia suggests infection. The cardiac examination may suggest congenital heart disease (e.g., critical coarctation of the aorta), and the pulmonary examination may reveal tachypnea, apnea, or respiratory distress suggestive of pneumonia or sepsis. The abdominal examination is important in an infant with a history of vomiting to exclude abdominal catastrophe or obstruction (e.g., atresia, volvulus, intussusception, or pyloric stenosis). The GU examination may reveal ambiguous genitalia suggestive of congenital adrenal hyperplasia (discussed later separately in the “Congenital Adrenal Hyperplasia [Adrenal Insufficiency]” section).
The most important diagnostic test in the ED for the neonate, infant, or child who is critically ill or shows altered mental status is a rapid bedside screen for serum glucose level. Confirm abnormal results with a venous sample sent to the laboratory. Although the treatment of hypoglycemia must be prompt and should not be delayed, the first blood sample taken from the hypoglycemic neonate or infant is critical for making a definitive diagnosis, and when possible, a gray-topped sample tube should be filled and placed on ice for additional studies, which may include serum insulin, C-peptide, growth hormone, cortisol, and glucagon levels.13
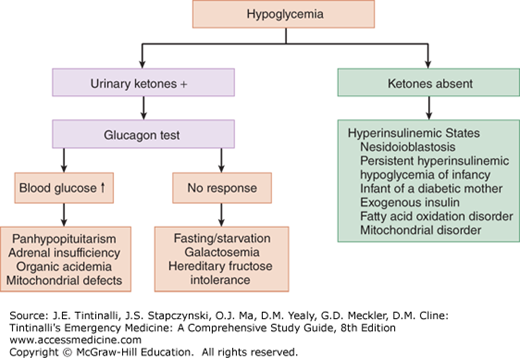
Evaluation of urine for ketones is the second important step. Ketonuria is characteristic of ketotic hypoglycemia, adrenal or growth hormone deficiency, and other inborn errors of metabolism. A lack of urinary ketones suggests hyperinsulinism or fatty acid oxidation defects.14,15 Serum insulin, C-peptide, and hormone analysis, as described above, can help differentiate among these diagnostic possibilities.
The administration of glucagon (0.3 milligram/kg IM or IV) in hypoglycemic states can be diagnostic and therapeutic. If glucagon is effective in normalizing serum glucose level, then the presence of hepatic stores is confirmed and the hypoglycemia is likely due to hormonal deficiency (panhypopituitarism or adrenal insufficiency) (Figure 144-1). Lack of response to glucagon suggests poor glycogen stores. Among nonresponders to glucagon, fasting is the most common cause, followed by galactosemia and hereditary fructose intolerance, although children with ketotic hypoglycemia often fail to respond to glucagon as well.
Additional laboratory evaluation is directed by the clinical picture and differential diagnosis and may include cultures of the blood, urine, and cerebrospinal fluid when sepsis is suspected. Definitive diagnosis of specific inborn errors of metabolism may require evaluation of levels of urine organic acids and serum amino acids and serum lactate (discussed later in “Inborn Errors of Metabolism”), as well as serum ammonia and lactate.
Routine imaging studies are not required for most cases of hypoglycemia, but may be useful if there are underlying organ anomalies.
Treat hypoglycemia promptly while awaiting diagnostic results. IV dextrose is the primary treatment and may be given enterally (PO, nasogastric tube) or parenterally (IV or IO). The dose of dextrose is 0.5 to 1.0 gram/kg regardless of the route of administration. Newborns should receive 5 mL/kg of 10% dextrose, whereas infants and children should receive 1 to 2 mL/kg of 25% dextrose. With adequate IV access, 1 mL/kg of 50% dextrose may be administered to older children as to adults. Some recommend a 0.2 gram/kg dextrose bolus to minimize hyperglycemia and resultant insulin secretion, which prolongs hypoglycemia. Use of dilute solutions in younger patients is suggested to minimize the vascular injury associated with more concentrated fluids.
Provide maintenance dextrose at a rate of 6 to 8 milligrams/kg/min with 10% dextrose, which is 1.5 times the normal maintenance rate for infants and children. If IV or IO access or nasogastric tube placement cannot readily be initiated, glucagon, 0.3 milligram/kg IM, may be given. Refractory hypoglycemia may be seen in hyperinsulinemic states such as insulin-secreting tumors and is suggested by hypoglycemia requiring administration of more than 6 to 8 milligrams/kg/min. Frequent reevaluation and titration of infused dextrose are necessary in this situation. If adrenal insufficiency is suspected, give hydrocortisone, 25 grams IV or IM for neonates and infants, 50 grams for toddlers and school-age children, and 100 grams for adolescents. The management of hypoglycemia is summarized in Table 144-1.
| Patient Age | Dextrose Bolus Dose | Dextrose Maintenance Dosage | Other Treatments to Consider |
|---|---|---|---|
| Neonate | D10 5 mL/kg PO/NG/IV/IO | 6 mL/kg/h D10 | Glucagon, 0.3 milligram/kg IM Hydrocortisone, 25 grams PO/IM/IV/IO |
| Infant | D10 5 mL/kg PO/NG/IV/IO or D25 2 mL/kg | 6 mL/kg/h D10 | Glucagon, 0.3 milligram/kg IM Hydrocortisone, 25 grams PO/IM/IV/IO |
| Child | D25 2 mL/kg PO/NG/IV/IO | 6 mL/kg/h D10 for the first 10 kg + 3 mL/kg/h for 11–20 kg + 1.5 mL/kg/h for each additional kg >20 kg | Glucagon, 0.3 milligram/kg/IM Hydrocortisone, 50 grams PO/IM/IV/IO |
| Adolescent | — | 6 mL/kg/h D10 for the first 10 kg + 3 mL/kg/h for 11–20 kg + 1.5 mL/kg/h for each additional kg >20 kg | Glucagon, 0.3 milligram/kg IM Hydrocortisone, 100 grams PO/IM/IV/IO |
Because sepsis is always in the differential diagnosis of the neonate, infant, or child who is critically ill or shows altered mental status, provide prompt broad-spectrum antibiotics as indicated by the clinical picture. This includes ampicillin, 50 milligrams/kg, and gentamicin, 5 to 7.5 milligrams/kg, or cefotaxime, 50 milligrams/kg, for neonates and infants in the first 2 months of life, and ceftriaxone, 50 milligrams/kg (100 milligrams/kg if meningitis is suspected), for older infants and children.
All neonates and infants with symptomatic hypoglycemia requiring ED resuscitation should be admitted to the hospital for further evaluation and treatment. Patients for whom sepsis is a concern and those requiring dextrose beyond the expected 6 to 8 milligrams/kg/h may require admission to the intensive care unit.
INBORN ERRORS OF METABOLISM
Although the diversity and complexity of inborn errors of metabolism in infants and children may seem overwhelming, keep in mind that making a definitive diagnosis is not as important as maintaining a high suspicion and that acute stabilization and management are relatively simple. As a group, these disorders involve enzyme deficiencies that lead to errors of metabolism resulting in the accumulation of various toxic biochemical products, which can cause dysfunction of multiple organ systems, especially the CNS. Although each individual type of inborn error of metabolism is extremely rare, as a group they are relatively common, with an incidence ranging from 1 in 1400 to 1 in 200,000 live births.13,14
Clinical manifestations of inborn errors of metabolism are a result of the accumulation of toxic metabolites and their effects on end-organs. Common symptoms and signs of inherited metabolic disorders include acute encephalopathy with or without metabolic acidosis and hypoglycemia (discussed earlier in “Hypoglycemia”). Because most metabolic toxins cross the placenta and are cleared by maternal enzymes, most newborns are asymptomatic and present after varying delays once enteral feeding begins. Hypoglycemia may be the primary presentation of some inherited disorders of metabolism and is discussed earlier (see “Hypoglycemia”). Jaundice and hepatic dysfunction can be seen in a number of inherited disorders such as galactosemia.16 Shock and cardiovascular collapse can occur with congenital adrenal insufficiency, but nonmetabolic conditions such as congenital heart disease and sepsis must be considered in the differential diagnosis of infants presenting in extremis. The discussion here is limited to conditions that typically present in early infancy with the potential for life-threatening consequences.
Most inborn errors of metabolism result from single-gene defects with a variety of inheritance patterns. The defects result in abnormal metabolism of protein, fat, carbohydrates, or other complex molecules. Affected proteins include enzymes, enzyme cofactors, and transport proteins. The result of these varied deficiencies is the accumulation of toxic substrates upstream of the impaired protein or of intermediates derived from alternate metabolic processes downstream. On the basis of metabolic and clinical manifestations, these disorders can often be grouped into those defects resulting in hyperammonemia, metabolic acidosis, hypoglycemia, or hyperbilirubinemia and liver dysfunction.16
Urea cycle defects, organic acidemias, and some fatty acid oxidation defects may result in the accumulation of ammonia, leading to encephalopathy.17
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


