and Richard A. Jaffe2
(1)
David Geffen School of Medicine at UCLA, Los Angeles, California, USA
(2)
Stanford University School of Medicine, Stanford, California, USA
Keywords
OpioidPharmacologyNormeperidineShiveringAt one time meperidine was the most commonly used opioid for surgical anesthesia or as a supplement to regional anesthesia. Its decline as an operating room analgesic after many years of successful use was the result of at least two events. One was the development of ultra short acting opiates. Fentanyl, synthesized in the late 1960s by Paul Janssen, and later its derivatives sufentanil, alfentanil, and remifentanil, gradually replaced meperidine in the operating rooms of United States hospitals. The general thinking was that shorter acting opiates would be easier and safer to administer and manage than longer acting ones. The other event was that meperidine became an unpopular analgesic drug among clinicians in general medicine and surgery because of its side effects, namely nausea and vomiting, adverse interaction with MAO inhibitor drugs, and normeperidine toxicity causing seizures. The increasing distain for meperidine within medical circles had a strong influence on the anesthesia community. However, the reasons that clinicians were abandoning meperidine were for the most part either specious or irrelevant to its use in clinical anesthesia. For example, there is no evidence that meperidine is more emetogenic than the newer opiates. Further, the MAO inhibitors have been almost completely replaced by more effective drugs that do not have any unique interaction with meperidine. Finally, there has never been a case report or study documenting seizures from the use of meperidine in the operating room. Nevertheless, today meperidine is used almost exclusively as a single dose only treatment for postoperative shivering or as an analgesic for short, outpatient diagnostic procedures. As a result, whole generations of anesthesia providers have not had training in the use of or experience with meperidine. This is very unfortunate because meperidine has properties that make it an excellent operating room analgesic.
History
Meperidine was the first opioid to be synthesized in the laboratory and in 1939 that was a unique way to create a new drug. In an effort to develop a better vagolytic drug than atropine, two German scientists, Von O. Eisleb and O. Schaumann reconfigured the atropine molecule, and developed several new agents, the most promising of which was one they named “dolantin” [1, 2]. They administered dolantin to both mice and cats and observed that it was a less potent vagolytic than atropine, but much to their surprise it had spasmolytic and analgesic effects. In comparative studies with morphine, they found it to be an effective but less potent analgesic. In comparative studies with codeine, they found that it was as effective as codeine in suppressing tracheal cough reflexes.
In 1943 Rovenstine and Batterman reported on the successful use of meperidine as a preanesthetic medication in 338 patients who were to undergo general anesthesia [3]. Shortly thereafter, Batterman and Himmelsbach reported in patients that meperidine 100 mg produced an analgesic effect equivalent to morphine 10 mg [4]. Following these reports, the drug was gradually introduced into operating room anesthesia as a substitute for morphine as part of a “balanced” anesthetic technique.
Pharmacology
Meperidine (1-methyl 4-phenyl-piperidine 4-carboxylic acid ethyl ester hydrochloride) is a member of the phenylpiperidine series , which includes fentanyl, sufentanil, alfentanil and remifentanil. Meperidine chemical structure is very similar to that of atropine and quite different from morphine (Fig. 10.1). As a result, it is not surprising that it has some of the same pharmacological effects as atropine. For example, it has a mild antimuscarinic effect manifested as a slight increase in heart rate. When initially studied, it was thought that meperidine had a mild spasmolytic effect on smooth muscle which would provide some suppression of coughing when the tracheal mucosa is irritated, as well as relaxing gastric and bowel smooth muscle thereby delaying gastric emptying and decreasing bowel motility. Because of its relaxant effect on airway smooth muscle, early investigators thought that it might be a useful drug for treating patients with bronchial asthma, but that did not prove to be the case. More recent studies have also shown that the spasmolytic effects on smooth muscle are minimal, and probably not of therapeutic benefit. However, it has two effects that are not characteristic of atropine, namely sedation and analgesia. Meperidine will produce mild sedation and euphoria , both of which make it a useful drug as a premedicant for patients undergoing regional anesthesia or diagnostic procedures under monitored anesthesia care. More importantly, meperidine is an effective analgesic because of its action on the μ receptors, and to a lesser extent on the δ and k receptors in the central nervous system . Because of its analgesic property, it was the most commonly used opiate in clinical anesthesia for more than 20 years until fentanyl was developed.
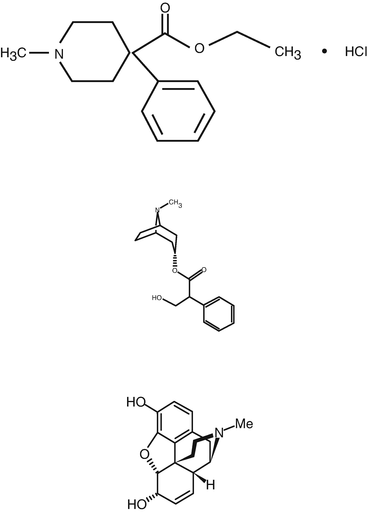
Fig. 10.1
Chemical structures of meperidine (top), atropine (center) and morphine (bottom)
Meperidine is somewhat less lipid soluble than fentanyl , so its onset of action following intravenous administration is about 5 min compared to 1–3 min for fentanyl. Meperidine duration of action from a single dose is about 2–3 h. Meperidine has a large volume of distribution and 65–75 % is protein bound. Meperidine undergoes relatively rapid metabolism in the liver via two routes, the predominant pathway being hydrolysis to form meperidinic acid, and the lesser pathway being N-demethylation to form the active metabolite normeperidine (Fig. 10.2). Normeperidine then undergoes hydrolysis in the liver to form normeperidinic acid. Both acid forms are inactive and are excreted by the kidneys. Animal studies have demonstrated that normeperidine has an analgesic potency about half of its parent compound. However, accumulation of normeperidine (elimination half-life 15–30 h) may cause central nervous system irritability as manifest by myoclonic jerks and seizures. Evidence that the same may occur in humans is entirely circumstantial, based on case reports of patients developing nervousness, dysphoria, tremors, myoclonus and occasionally seizures after having received meperidine for days or weeks as a treatment for chronic pain. However, there is no clear relationship between normeperidine toxicity and CNS irritability and seizures in patients. Manifestations of toxicity are most likely to occur in patients receiving meperidine in high doses (>1000 mg/24 h), for prolonged periods (days to weeks), or those patients in renal failure, which may prolong the normal renal excretion of normeperidine. Normeperidine toxicity has never been an issue with the use of meperidine in operating room anesthesia because the doses of meperidine used as part of a balanced anesthesia technique are too low (less than 300–400 mg) and too short a duration of administration (much less than 24 h) to produce normeperidine neurotoxicity even in patients in renal failure. There has never been a documented case of a seizure due to meperidine when administered as an adjuvant in clinical anesthesia.
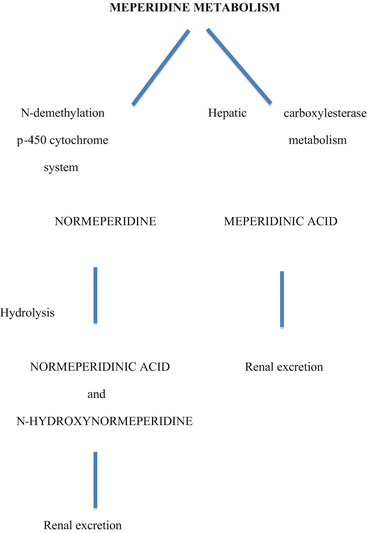
Fig. 10.2
Meperidine metabolism
In some patients, meperidine may cause a histamine reaction , most commonly manifested as cutaneous redness and blotching around the vein at the site of an intravenous injection. This may cause some itching specifically at the injection site. Nasal itching is also common, but it is not known whether the mechanism is histamine release or a central mechanism. These are trivial events, do not require treatment, and subside in a few minutes. It has been suggested that meperidine is associated with significant histamine release, and should be used cautiously in patients with a history of asthma [5, 6]. However, the authors did not provide any definition of “significant”, or provide any case reports documenting an asthmatic event due to meperidine. In our extensive use of meperidine, the histamine reactions have always been local, never systemic. This is another example of how an undocumented postulate becomes “fact” and alters medical care.
Usually there are no cardiovascular changes associated with intravenous meperidine injections unless a very large dose (100 mg) is given as a single bolus. The hypotension from such large doses is probably due to myocardial depression with or without some vascular dilatation from histamine release.
Respiratory depression is a feature of all opiates, and meperidine is no exception. The respiratory depression is dose related and is no worse or better than other opiates. With increasing doses, meperidine causes a progressive decrease in respiratory rate and increase in tidal volume, with the net effect being a decrease in minute and alveolar ventilation and increasing arterial and end-tidal carbon dioxide. If used in combination with a volatile anesthetic such as sevoflurane or isoflurane, tidal volume decreases instead of increasing, so alveolar ventilation is decreased even more. However, unless given in very large bolus doses, meperidine will not cause cessation of breathing or apnea. In contrast to meperidine, fentanyl and its derivatives may cause apnea even in small doses (50–100 μg) and even in healthy patients. This difference has important implications in the selection of an opiate as a premedicant or for treatment of acute postoperative pain in the postanesthesia care unit. For example, orthopedic patients who come to hospital for surgery are often in pain and accustomed to taking one or more of a variety of analgesics. Usually they are instructed not to take any analgesics the day of surgery so they arrive in the preoperative suite complaining of pain. Nurses in the unit then call the anesthesia provider requesting an order for an analgesic. Meperidine is an ideal drug in this circumstance. A dose of 20–25 mg can be administered safely intravenously and it will alleviate the pain within 10 min. If after 15 min the patient is still complaining (probably because of opiate tolerance), a second, similar dose can be administered safely. Doses administered in this manner will not make the patient apneic. The same drug scenario can be used for patients experiencing acute pain in the postanesthesia care unit. Many anesthesia providers would use fentanyl in these circumstances, but we would strongly recommend against it. Fentanyl may cause sudden apnea, and if not detected immediately may cause hypoxemia, hypotension and even cardiac arrest. We would urge that anesthesia providers not order fentanyl or its derivatives in any setting other than those where the provider can watch the patient for at least 15–20 min after its administration. We know of a case where fentanyl was administered to a patient in pain, put into a CT scanner, and when removed from the scanner the patient was dead, or where emergency room patients were given fentanyl to reduce a fracture or suture a wound, and when they returned to do the procedure, the patients were dead. Likewise, we have seen multiple cases of acute apnea following administration of fentanyl in a recovery room, but fortunately none of them resulted in permanent injury.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


