Figure 1.1. Typical structures of local ester and amine anesthetic molecules.
where
pH is the solution or tissue pH and
pKa is the pH at which half the local anesthetic molecules are in the base form and half in the acid form.
The value for pKa is unique for any local anesthetic and is a measure of the tendency for the molecule to accept a proton when in the base form or to donate a proton when in the acid form. Most local anesthetics have a pKa between 7.5 and 9.0.
Because local anesthetics are supplied as unbuffered acidic solutions (pH = 3.5–5.0), there are approximately 1,000 to 100,000 times more molecules in the charged form than the uncharged form (which helps to keep the local anesthetic in solution). Because extracellular tissue pH is approximately 7.4, the proportion of molecules in the charged form decreases by a factor of somewhere between 500 and 10,000 when injected into tissue. For example, because mepivacaine has a pKa of 7.6, there would be 1,000 times as many molecules in the protonated form (weak acid) than in the uncharged form in a commercially supplied solution at pH 4.6. Once injected into tissue with a pH of 7.4, many of the charged mepivacaine molecules would “donate” their protons so that only approximately 1.6 times as many will be charged as uncharged. As discussed in Section IV.E, it is critical to local anesthetic action that they are capable of transitioning between the charged and uncharged forms.
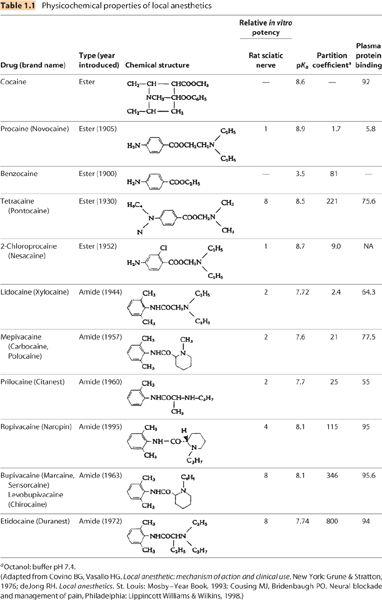
B. Hydrophobicity (Table 1.1). Local anesthetics vary in the degree to which they dissolve in aqueous (hydrophilic) versus lipid (hydrophobic) environments. Differences in hydrophobicity are primarily the result of differences in the types of chemical groups bound to the tertiary amine (Figure 1.1). The charged form of any individual local anesthetic is more hydrophilic than is the corresponding uncharged form. Hydrophobic character is often, and inaccurately, referred to as lipid solubility. Greater hydrophobicity correlates with greater local anesthetic potency and duration of action (see Section V.A and Chapter 2).
1. Hydrophobicity is determined by adding the local anesthetic to a vessel containing two immiscible liquids—an aqueous buffer and a hydrophobic “lipid.” Lipids are usually chosen in an effort to mimic the hydrophobic character of cellular lipid membranes; octanol, olive oil, and n-heptane are commonly used lipids. The local anesthetic is added to the vessel and the vessel is agitated to “mix” the two liquids. The solution is allowed to sit and the liquid phases to separate. After separation, the concentration of local anesthetic is measured in the aqueous phase and in the lipid phase. The resultant ratio of the concentrations is the “distribution coefficient,” which is often inappropriately simplified as the “lipid solubility.”
2. Importantly, the distribution coefficient so determined will vary greatly depending on:
a. The pH of the aqueous phase because this will determine what percentage of the local anesthetic is charged (more hydrophobic) or uncharged (more hydrophilic). A pH of 7.4 is common and the resulting distribution coefficient is termed the partition coefficient. The distribution coefficient is commonly measured using the local anesthetic base and an aqueous phase pH significantly above the drug’s pKa, so all of the local anesthetic is effectively uncharged.
b. The lipid used. Different lipids will yield very different distribution coefficients and the values determined in one solvent system cannot be compared with those determined in a different system. Referring to a drug’s “lipid solubility” without defining the system in which it was determined is incomplete information.
c. The form of the local anesthetic (i.e., base or salt). Consequently, tables that simply list a local anesthetic’s “lipid solubility” without information as to how it was determined are not particularly useful. In Table 1.1, local anesthetic partition coefficients are reported for chloride salts of local anesthetics in octanol and buffer at pH 7.4 (octanol: buffer7.4).
C. Protein binding. Binding to plasma proteins varies between 5% and 95% (Table 1.1). In general, more hydrophobic drugs have higher protein binding. In fact, properties sometimes attributed to a drug’s degree of “protein binding” are probably actually related to their hydrophobicity. Whether plasma protein binding has any relationship to tissue protein binding is unknown and should not be assumed.
1. α1-Acid glycoprotein and albumin are the primary plasma proteins to which local anesthetics bind. Binding to these proteins is pH dependent and binding decreases during acidosis, because the number of available binding sites decreases in an acidic environment.
2. In plasma, it is the unbound or “free” fraction of local anesthetic that is capable of leaving plasma to enter organs like the brain or heart. Consequently, it is the free fraction that is responsible for systemic toxicity.
a. Patients with low plasma protein concentrations (e.g., malnutrition, cirrhosis, and nephrotic syndrome) are at greater risk of systemic toxicity than are patients with normal plasma protein concentrations and patients with high plasma protein concentrations (e.g., some cancers) are afforded a degree of protection (1).
IV. The sodium channel and nerve conduction
A. Sodium channel structure (Figure 1.2). The mammalian sodium channel is a transmembrane protein composed of three subunits that form a voltage-sensitive, sodium-selective channel through the neuronal membrane. To date, ten distinct human genes coding for ten structurally different sodium channels have been identified. Different isoforms are expressed in different tissues (e.g., muscle, heart, central nervous system, and peripheral nervous system). It is possible that there are mutations that confer either increased or decreased sensitivity to local anesthetics [in fact, such induced mutations have been produced in experimental systems (2,3)], but to date none have been identified clinically.
B. Conduction. At rest, neurons maintain an electrochemical gradient across their membranes because Na+/K+-ATPase (adenosine triphosphatase) pumps three Na+ ions out of the axoplasm for every two K+ ions pumped in. Consequently, the axon interior is relatively negative (–50 to –90 mV) and sodium poor compared to the exterior (Figure 1.2). When the nerve is sufficiently “stimulated,” sodium channels in a very localized region of the nerve membrane open thereby permitting Na+ ions to move down their electrochemical gradient into the axon interior and locally “depolarize” the axonal membrane. If the magnitude of the depolarization exceeds “threshold” (i.e., the transmembrane potential decreases sufficiently), then sodium channels in the adjacent membrane are induced to open (this is what is meant by “voltage-sensitive”) which in turn depolarizes even more membrane areas and induces even more distant sodium channels to open. In this way, the depolarization spreads down the axonal membrane producing an action potential.
C. Repolarization. After a few milliseconds, the sodium channel is inactivated by a time-dependent conformation change that closes an inactivation gate (Figure 1.2). In the inactivated state, the sodium channel cannot conduct Na+ and cannot be reopened if stimulated (analogous to the cardiac refractory period). Initially, resting membrane potential recovers toward normal by the extracellular movement of K+ and later by Na+/K+ exchange by ATPase. As the resting membrane potential is restored, the sodium channel undergoes additional conformation changes to enter the closed (resting) state during which it does not conduct Na+ ions, but a sufficient stimulus (e.g., depolarization, sensory transduction, neurotransmitter binding) will convert the channel to the open state. Importantly, the binding affinity of local anesthetics varies with the state of the sodium channel, being greatest in the inactivated state and least in the resting (closed) state. These state-dependent differences in binding affinity underlie “phasic” or “rate-dependent” block (see Section V.B). Also, differences between local anesthetics in the degree to which they exhibit state-dependent differences in binding affinity underlie the differences in their relative cardiovascular toxicity (see Chapter 3).
D. Local anesthetic binding. There is no “receptor” for local anesthetics; rather there is a “binding site.” Directed mutagenesis studies indicate that the local anesthetic binding site is located within the sodium channel near its intracellular opening (Figure 1.2) (2). Local anesthetics block action potentials by preventing Na+ movement through the sodium channel; either by physically blocking Na+ or by preventing a necessary change in sodium channel conformation that would permit Na+ to traverse the pore.
1. The local anesthetic binding site consists of a hydrophobic region to which the hydrophobic portion of the local anesthetic molecule is assumed to interact and a hydrophilic region where the quaternary amine interacts (Figure 1.2). Amino acid substitutions at these sites prevent local anesthetics from being effective.
E. Model of local anesthetic action. In vitro experiments using giant squid axon have shown that permanently charged quaternary amine local anesthetics have relatively weak local anesthetic activity when applied outside the nerve membrane, but are quite potent when inserted directly into the nerve cytoplasm. Conversely, uncharged tertiary amine local anesthetics applied intraneurally are not very effective local anesthetics. These observations lead to the following model for tertiary amine local anesthetics (Figure 1.3).
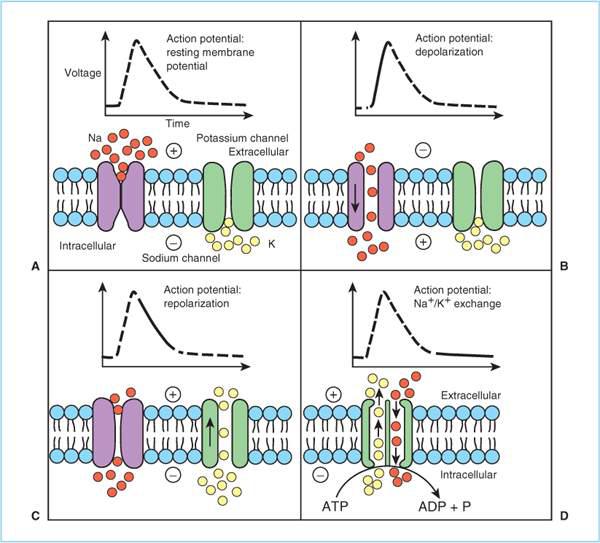
Figure 1.2. Sodium and potassium channel function and ion movements during nerve depolarization. A: At rest, the sodium channel is in the closed confirmation and there is a relative excess of sodium ions (solid circles) in the extracellular space and a relative excess of potassium ions in the intracellular space (open circles). Because there are approximately three positively charged sodium ions in the extracellular space for every two charged sodium–potassium ions in the intracellular space, the intracellular space is negative (−50 to −90 mV) relative to the extracellular space. B: Following a sufficient stimulus, the voltage-gated sodium channel confirmation changes to the open configuration, and sodium ions flow down their electrochemical gradient into the interior of the neuron, resulting in depolarization. C: At the peak of the action potential, the sodium channel conformation changes spontaneously to the inactivated state, which prevents further sodium entry and is refractory to reopening in response to a stimulus. Simultaneously, the voltage-gated potassium channels open, and potassium flows down its concentration gradient to render the neuron interior negative relative to the exterior (repolarization). D: The sodium–potassium pump (Na+/K+ adenosine triphosphatase [ATPase]) exchanges three intracellular sodium molecules for every two extracellular potassium molecules, thereby restoring the resting membrane potential and moving the sodium channel to the closed confirmation. ADP, adenosine diphosphate; ATP, adenosine diphosphate; P, phosphate. (Adapted from Baras K, Clitten S. Clinical Anesthesia, 3rd edition.)
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


