Diagram of multivesicular liposomal bupivacaine.
Body heat destabilizes the phospholipid layer causing degradation of the membrane, resulting in a slow controlled release of bupivacaine, typically over 72 to 96 hours (Figure 5.2). Plasma concentrations show two distinct peaks: one between 0 and 2 hours due to uptake of the 3% extra-liposomal bupivacaine and a second, more significant, peak between 24 and 48 hours due to gradual sustained release of bupivacaine [1]. For perspective, free bupivacaine (0.5%) reaches its plasma peak at 37.5 minutes [2].
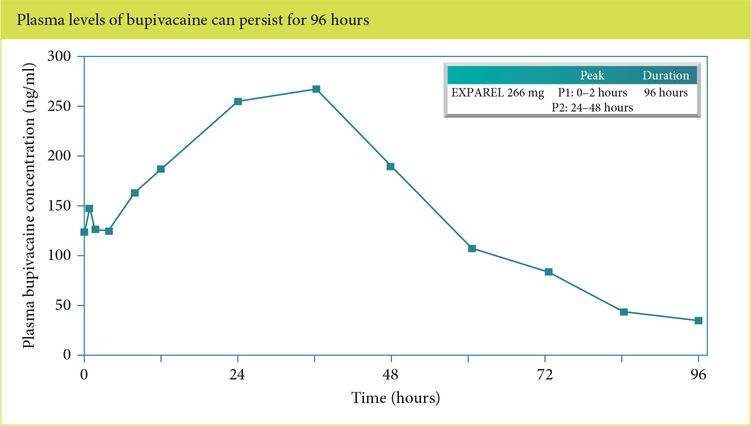
Bimodal peak concentration with liposomal bupivacaine infiltration.
The pharmacokinetic effects of liposomal bupivacaine are related to release of active drug from the liposomal matrix. Richard et al. [3] compared multivesicular liposomes in concentrations of 9, 18, and 30 mg/kg with 9 mg/kg plain bupivacaine injected by wound infiltration in rabbits. Resulting Cmax (peak serum concentration) was dose dependent and found to be between 107 ± 27.6 and 307 ± 148 ng/ml, which is much lower than that of plain bupivacaine (620 ± 89.9 ng/ml). Davidson et al. [2] compared subcutaneous injection of 20 ml of 2% liposomal bupivacaine vs. 20 ml of 0.5% plain bupivacaine in human subjects. No statistical difference was found in Cmax; however, there was an almost ten-fold increase in terminal half-life (131 min vs. 1,294 min) in plain and liposomal, respectively.
After release and systemic uptake of liposomal bupivacaine, metabolism and excretion are by the liver and kidney, respectively. Similar to free bupivacaine and other amides, liposomal bupivacaine is metabolized in the liver via conjugation, N-dealkylation, and hydroxylation [4]. The major metabolite is pipecoloxylidine, which is largely inactive [5]. Elimination and excretion of metabolites and conjugation byproducts is via the kidneys. Minimal unmetabolized bupivacaine is excreted via the renal system. Thus, liposomal bupivacaine has an extended terminal half-life with predictable peak concentration times without active metabolites.
2. Discuss the efficacy of the medication versus placebo and other local anesthetics
Several randomized clinical trials have examined the efficacy of liposomal bupivacaine compared both with placebo and with free bupivacaine. The original two trials compared liposomal bupivacaine to placebo in patients undergoing bunionectomy and hemorrhoidectomy [6–7]. These studies found reduced pain scores, lower opioid consumption, and longer time to first opioid administration. However, while the primary parameter was area under the curve (AUC), a calculation used by the developers of liposomal bupivacaine to describe intensity of pain occurring during the first 72 hours; clinically, significant differences were present only in the first 24 hours (Figures 5.3 and 5.4). It should also be noted that in the liposomal bupivacaine arms of the studies, a multimodal analgesic protocol was also employed; conversely, the placebo group only received standard opioid rescue medications.
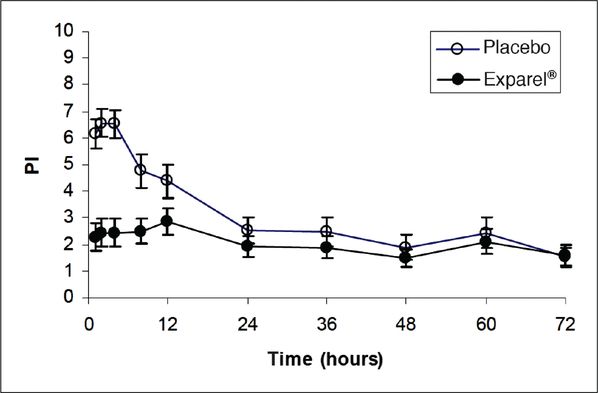
Mean pain intensity vs. time plot for hemorrhoidectomy study – showing minimal difference in effect on pain measures compared with placebo after 24 hours.
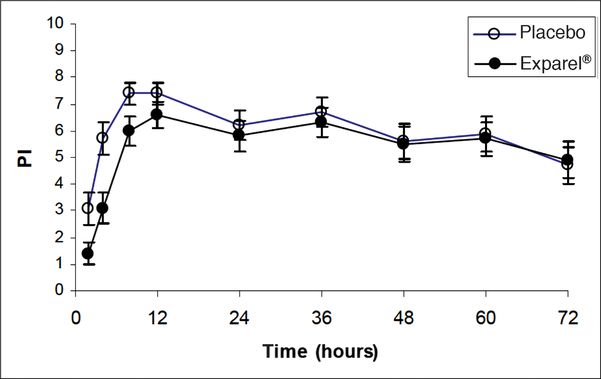
Mean pain intensity vs. time plot for bunionectomy study – showing minimal difference in effect on pain measures compared with placebo after 24 hours.
A trial directly comparing free bupivacaine with epinephrine to liposomal bupivacaine involved patients undergoing hemorrhoidectomy with 101 patients randomized to 300 mg liposomal bupivacaine and 103 receiving 100 mg bupivacaine plus 1:200,000 epinephrine. This trial’s results have not been formally published, but a thorough summary is available beginning on page 126 of an FDA report [8]. The results of this trial were equivocal, with an AUC of the numerical rating scale pain score at rest (NRS-R) at 96 hours that was not different between liposomal bupivacaine and free bupivacaine with epinephrine, respectively (396 ± 213 vs. 359 ± 194, P = 0.15).
Several other earlier phase II trials summarized by Bergese et al. show minimal differences in patients avoiding opioid rescue and statistically insignificant differences in pain scores [9]. This review included the use of liposomal bupivacaine in various procedures including inguinal hernia repair, hemorrhoidectomy, breast augmentation, and total knee arthroplasty (TKA).
For patients undergoing TKA, a phase II dosing study compared 150 mg of bupivacaine HCl with liposomal bupivacaine (133 mg, 266 mg, 399 mg, and 532 mg) [10]. There were no differences in the AUC of the NRS-R in patients receiving clinically acceptable doses of liposomal bupivacaine. A small yet statistically significant difference was found between the bupivacaine HCl and liposomal bupivacaine 532 mg group; however, this dose is in excess of the FDA published maximum dose of 266 mg of liposomal bupivacaine.
In contrast, in a meta-analysis of multiple surgical procedures, Dasta et al. [11] reported a significant decrease in AUC at 72 hours, increased median time to first opioid use (10 hours vs. 3 hours, P < 0.0001), decreased amount of total opioid used, and decreased incidence of opioid-related adverse effects for liposomal bupivacaine.
3. Analyze the safety profile of liposomal bupivacaine
The most common adverse reactions reported during preliminary clinical trials involving liposomal bupivacaine were non-specific symptoms such as nausea, constipation, and vomiting [5]. Across a large pool of 21 clinical studies involving a total of 823 patients exposed to liposomal bupivacaine, most adverse events were classified as mild to moderate and unrelated to the study drug itself. In those studies directly comparing both liposomal and free bupivacaine, no clinically relevant differences in frequency or severity of adverse effects were observed. The Incidence of serious adverse events was actually lower among liposomal bupivacaine recipients when compared to free bupivacaine recipients (1.9% vs. 3.9%, respectively).
A review by Baxter et al. included ten clinical trials with various doses of liposomal bupivacaine and bupivacaine HCl and showed minimal statistically significant differences in outcomes such as patient satisfaction, wound scarring, and incidence of erythema [12]. No evidence of adverse clinical impact on wound or bone healing was seen across the various studies included. Further, the total incidence of adverse effects associated with local anesthetics was equivocal between similar doses of bupivacaine HCl and liposomal bupivacaine (8–19% and 9–20%, respectively).
Cardiac-related adverse effects associated with liposomal bupivacaine (and free bupivacaine) are tachycardia, bradycardia, palpitations, and extra systoles. These occur at incident rates of less than 2% [13]. QT-specific studies indicate minimal if any changes in the QT interval at doses of up to 750 mg administered subcutaneously in healthy volunteers [14].
There are potential drug interactions to consider when administering liposomal bupivacaine. Direct (or indirect) contact with certain drugs can cause a rapid increase of free bupivacaine, resulting in altered liposomal bupivacaine characteristics. Specifically, mixing liposomal bupivacaine with local anesthetics with different lipid solubilities can cause immediate release of free bupivacaine from encapsulated bupivacaine if given together locally. Further, co-administration of free bupivacaine (while not dramatically altering pharmacokinetic properties) will increase overall exposure to bupivacaine.
A patient safety concern is the milky-white appearance of liposomal bupivacaine that is very similar to propofol. Numerous efforts have been made to increase the product safety in terms in packaging and labeling [15], such as different-sized vials and distinctive fonts. However, very little visual distinction between the two products remains when they are pulled into syringes, creating the potential for an inadvertent intravenous administration and resultant local anesthetic systemic toxicity (LAST) [16]. Because current FDA indications only include wound infiltration, the administration of liposomal bupivacaine is likely to take place on the surgical field. Another potential inadvertent drug substitution also on the surgical field may be with Surgifoam® (Ethicon) or GelFoam® (Pfizer).
Finally, product storage is an important consideration due to the temperature-sensitive characteristics of liposomal bupivacaine. Recommended storage temperatures are between 2°C and 8°C (refrigerated). It should not be frozen or exposed to high temperatures (>40°C). A temperature indicator is present on the product insert, which turns from green to white when exposed to frozen temperatures. When in unopened vials stored at 20°C to 25°C, the medication remains stable for up to 30 days. Re-refrigeration is not recommended [17].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


