Chapter 9
Hemorrhagic Shock
Cardiogenic shock (see Chapter 8) results from an inability of the heart to effectively pump blood to surrounding tissues. Compensatory activation of the sympathetic nervous system increases systemic vascular resistance in an attempt to restore perfusion pressure that in some patients is manifested by cool extremities with a netlike pattern of bluish skin mottling known as livedo reticularis.
Distributive shock (Chapter 10) is characterized by a loss of vasomotor tone in capacitance and resistance vessels. Therefore, circulating blood volume is effectively insufficient (“relative hypovolemia”). In addition, it results in a low afterload state as a result of low systemic vascular resistance.
Hypovolemic shock results from a decrease in actual intravascular volume, the etiology of which may be myriad (Box 9.1). Hemorrhagic shock, the most common form of hypovolemic shock, is classically divided into four stages of severity (Table 9.1). These four stages correspond to the progression of blood loss and the associated physiologic responses in otherwise healthy individuals with normal cardiopulmonary systems.
TABLE 9.1
Estimated Blood Loss Based on Patient’s Initial Presentation

∗Values are based on an adult with a predicted body weight (PBW) of 70 kg in which total intravascular blood volume is estimated at 70 mL per kg PBW or, in this example, 70 mL x 70 kg PBW = 4900 mL or ~5000 mL.
From American College of Surgeons: Advanced Trauma Life Support Student Course Manual, 8th Ed. Chicago: American College of Surgeons, 2008, with permission.
Although conceptually useful, the distinction between cardiogenic, distributive, and hypovolemic shock is somewhat artificial as shock is frequently multifactorial. For instance, a patient who has sustained severe thermal burns will almost certainly manifest hypovolemia as intravascular volume migrates into the interstitial space secondary to capillary leak. The same patient may also have attenuated vasomotor tone secondary to the systemic inflammatory response syndrome (SIRS; see Chapter 10) causing a distributive shock. Finally, a fraction of patients with distributive shock caused by sepsis may also have an element of cardiogenic shock resulting from the depression of myocardial function from circulating inflammatory mediators or from preexisting disease or other factors.
Pathophysiology of Decreased Preload
Cardiac output is the product of heart rate and stroke volume. Stroke volume, in turn, is determined by ventricular preload, contractility, and afterload (Figures 8.1–8.4, Chapter 8). Preload corresponds to the stretch placed on cardiac muscle immediately prior to contraction. There is a direct relationship between sarcomere length (or stretch) and contractile force. As illustrated by the Starling curves, increasing preload increases the force of muscle fiber contraction and cardiac stroke volume up to a maximum after which the output plateaus (see Figure 8.1). The term preload is most accurately reflected by left ventricular end-diastolic volume (LVEDV) rather than the left ventricular end-diastolic pressure (LVEDP), which is commonly estimated in the intensive care unit (ICU) using the pulmonary artery catheter. For clinical purposes, the LVEDP is often assumed to be proportional to the LVEDV, although this relationship may become nonlinear, particularly in the noncompliant myocardium because of preexisting diastolic dysfunction (such as that caused by chronic hypertension) or ischemia. Preload is a function of the global circulating blood volume as well as venous return to the heart (see Box 9.1).
Physiologic and Pathophysiologic Changes in Hypovolemic Shock
Hypovolemic shock is characterized by a decreased cardiac preload, which results in a decreased stroke volume. Compensatory mechanisms for low cardiac output or hypotension are mediated by means of a sympathetic adrenergic response. In an attempt to maintain cardiac output, the force of cardiac contraction (inotropy) and the rate of contractility (chronotropy) both increase (see Figure 8.2). As hypovolemia progresses, in an effort to maintain an adequate perfusion pressure to organs, systemic vascular resistance and left ventricular afterload increase, redirecting blood flow from the periphery (skin, skeletal muscles, and fat in extremities) and from the splanchnic bed to the central circulation. For example, blood flow to the kidneys may decrease to only 5% to 10% of normal during acute hypovolemia, supporting the utility of monitoring urine output per hour as a gauge of adequate renal blood flow.
When all these compensatory mechanisms are active, the patient may tolerate even severe fluid loss with minimal or no tissue dysfunction (“compensated shock”) or with some reversible tissue dysfunction (“progressive shock”). In these states, resuscitation alone will restore the intravascular volume and is likely to reverse inadequate tissue perfusion. As both the volume of blood lost and length of time in shock increase, the degree of reversibility in response to intravascular volume resuscitation decreases and eventually it reaches an irreversible state in which survival is unlikely.
From a macrocirculatory standpoint, circulatory shock can be described as an imbalance between tissue oxygen supply and demand. Systemic oxygen delivery (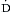 o2) is equal to the product of arterial oxygen content and cardiac output (Box 9.2). Oxygen consumption per minute (
o2) is equal to the product of arterial oxygen content and cardiac output (Box 9.2). Oxygen consumption per minute (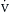 o2) is dependent on the body’s total metabolic activity, distribution of blood flow, and the ability of tissues to extract and utilize oxygen. The mixed venous oxygen saturation (S
o2) is dependent on the body’s total metabolic activity, distribution of blood flow, and the ability of tissues to extract and utilize oxygen. The mixed venous oxygen saturation (S o2) is measured in the pulmonary artery and is dependent on the relationship between
o2) is measured in the pulmonary artery and is dependent on the relationship between 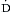 o2 and
o2 and 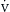 o2. The oxygen extraction ratio (
o2. The oxygen extraction ratio (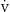 o2/
o2/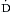 o2) represents the proportion of delivered oxygen to the oxygen consumed by the tissues. Under normal conditions, the
o2) represents the proportion of delivered oxygen to the oxygen consumed by the tissues. Under normal conditions, the 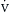 o2/
o2/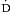 o2 is approximately 1/4, which corresponds to an S
o2 is approximately 1/4, which corresponds to an S o2 of ~75%. Under normal conditions, the amount of oxygen delivered to the tissues (
o2 of ~75%. Under normal conditions, the amount of oxygen delivered to the tissues (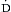 o2) is far in excess of oxygen consumption (
o2) is far in excess of oxygen consumption (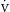 o2), and this explains why
o2), and this explains why 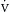 o2 typically varies independent of
o2 typically varies independent of 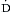 o2 (Figure 9.1).
o2 (Figure 9.1).
BOX 9.2 Basic Equations Related to Oxygen Delivery and Uptake

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


