HEMATOLOGIC EMERGENCIES
STACY E. CROTEAU, MD, MMS, ERIC W. FLEEGLER, MD, MPH, AND MARISA BRETT-FLEEGLER, MD
GOALS OF EMERGENCY THERAPY
The management of pediatric hematologic emergencies is directed toward immediate stabilization of the acutely ill patient to prevent morbidity and mortality related to severe anemia, infection, bleeding, and thrombosis. Patients at risk for infection and sepsis, such as neutropenic or asplenic patients, require prompt attention; quality metrics focus on time to initiation of antibiotic therapy for these patients. Simultaneously, significant hematologic findings of unknown etiology require thorough evaluation, especially where necessary therapies may obscure the underlying diagnosis or impede later diagnostic testing.
Hematologic emergencies arise in children who have been previously well, who have known blood disorders, or who have systemic disease. Initial measures of support, diagnosis, and treatment are based on general principles that often do not require a definitive diagnosis to initiate management.
KEY POINTS
 Hemoglobin levels may be normal in acute blood loss.
Hemoglobin levels may be normal in acute blood loss.
 The decision to transfuse red blood cells (pRBCs) should be based on etiology, severity, and chronicity of the anemia as well as on clinical symptoms and end-organ perfusion rather than hemoglobin values.
The decision to transfuse red blood cells (pRBCs) should be based on etiology, severity, and chronicity of the anemia as well as on clinical symptoms and end-organ perfusion rather than hemoglobin values.
 Patients with sickle cell disease require prompt evaluation for complications including stroke, acute chest syndrome, splenic sequestration, infection, vasoocclusive episodes, and priapism.
Patients with sickle cell disease require prompt evaluation for complications including stroke, acute chest syndrome, splenic sequestration, infection, vasoocclusive episodes, and priapism.
 Prompt evaluation and treatment of neutropenic patients is essential to decrease morbidity and mortality associated with infection. Appropriate cultures should be obtained but should not delay empiric antibiotic treatment. Disposition should be based on the underlying etiology of the neutropenia and clinical presentation.
Prompt evaluation and treatment of neutropenic patients is essential to decrease morbidity and mortality associated with infection. Appropriate cultures should be obtained but should not delay empiric antibiotic treatment. Disposition should be based on the underlying etiology of the neutropenia and clinical presentation.
 Increasingly, the management of immune thrombocytopenia (ITP) is guided by bleeding symptoms rather than platelet count.
Increasingly, the management of immune thrombocytopenia (ITP) is guided by bleeding symptoms rather than platelet count.
 Platelet disorders and von Willebrand disease (vWD) typically result in mucosal-type bleeding whereas hemophilia causes hemarthrosis and deep muscle bleeds. Drugs that interfere with platelet function (e.g., aspirin, nonsteroidal anti-inflammatory agents) should be avoided in patients with hemostatic defects.
Platelet disorders and von Willebrand disease (vWD) typically result in mucosal-type bleeding whereas hemophilia causes hemarthrosis and deep muscle bleeds. Drugs that interfere with platelet function (e.g., aspirin, nonsteroidal anti-inflammatory agents) should be avoided in patients with hemostatic defects.
RELATED CHAPTERS
Resuscitation and Stabilization
• Approach to the Injured Child: Chapter 2
Signs and Symptoms
• Gastrointestinal Bleeding: Chapter 28
Clinical Pathways
Medical Emergencies
• Gastrointestinal Emergencies: Chapter 99
• Oncologic Emergencies: Chapter 106
Trauma
• Abdominal Trauma: Chapter 111
• Thoracic Trauma: Chapter 123
Surgical Emergencies
• Abdominal Emergencies: Chapter 124
ANEMIA
Goals of Treatment
Severe anemia requires rapid evaluation and treatment to prevent hypoxia, congestive heart failure, end-organ damage, and death. When the etiology of the anemia is not immediately apparent, management focuses simultaneously on diagnostic and therapeutic interventions. Many management strategies for the anemic child are similar regardless of etiology, but special considerations are necessary in the setting of a destructive red cell process.
CLINICAL PEARLS AND PITFALLS
• The stabilization of the severely anemic child is guided by the clinical presentation more than by laboratory values. In chronic blood loss, low hemoglobin levels may be relatively well tolerated due to compensatory mechanisms and should not be the sole indication for transfusion.
• The acuity or chronicity of a clinical condition will impact the clinical status and thereby affect management. Prompt transfusions and isotonic fluid resuscitation needed for acute blood loss may be detrimental in chronically anemic patients.
• Transfusion risks versus benefits must be considered based on the clinical situation.
TABLE 101.1
CLASSIFICATION OF ANEMIA BY RED BLOOD CELL SIZE

Current Evidence
Anemia in the pediatric patient ranges from an incidental finding in an asymptomatic patient to acute or chronic processes presenting in a critically ill patient. The classification of causes of anemia according to (i) blood loss, (ii) increased RBC destruction, and (iii) decreased RBC production provides a framework for the evaluation of the anemic child.
Alternatively, comparing the MCV level with age appropriate normal values allows for classification of anemia as macrocytic, normocytic, or microcytic (see Table 101.1). A reticulocyte count indicates the reactivity of the marrow, and elevated levels are often found in blood loss and hemolysis. Decreased levels of serum haptoglobin, elevated lactate dehydrogenase, and increased unconjugated bilirubin along with the presence of heme in the urine suggest hemolysis.
BLOOD LOSS
CLINICAL PEARLS AND PITFALLS
• In acute hemorrhage, measured hemoglobin changes may lag behind blood loss, and normal values should not provide reassurance against clinically significant blood loss.
Clinical Considerations
Clinical Recognition
Anemia due to blood loss occurs from a variety of causes (Fig. 101.1). Overall, these conditions are divided into traumatic or atraumatic bleeding. The possibility of occult nonaccidental trauma must always be considered, particularly in younger children. Gastrointestinal hemorrhage is the most common cause of atraumatic blood loss, but postsurgical (e.g., posttonsillectomy hemorrhage), renal, gynecologic, and other etiologies may also present. In some cases, an anatomic lesion or process may combine with a congenital or acquired coagulopathy to precipitate significant anemia. For example, adolescent girls with unrecognized von Willebrand disease (vWD) may present with anemia due to both acute and chronic blood loss during menses.
Assessment for anemia should be considered in any patient with pallor, jaundice, or unexplained tachycardia. Asymptomatic chronic anemia may be detected as an incidental finding that requires further evaluation.
Triage
In the patient with known or suspected anemia due to blood loss, hypotension, hypoxia, or evidence of end-organ dysfunction is a medical emergency and warrants immediate intervention to prevent progression to cardiopulmonary collapse. Patients with both acute and chronic blood loss may become unstable. The chronicity of symptoms should not reassure the clinician, as it may be the exhaustion of physiologic compensatory mechanisms that prompts the patient to present to medical attention.
Initial Assessment
Initial assessment of a patient presenting with anemia secondary to blood loss includes a focused history targeted at potential etiology and symptoms of compromise related to anemia/hypoxia. In suspected blood loss, the clinician must assess for evidence of trauma including nonaccidental injury, postprocedure bleeding, symptoms of upper or lower gastrointestinal bleeding, medication use that could precipitate gastrointestinal bleeding, reports of epistaxis, hematuria or menorrhagia, and any concern for complications of pregnancy or hemorrhagic ovarian cyst. History related to symptomatic anemia should include fatigue, exercise intolerance, syncope, orthostasis, chest pain or dyspnea, decreased urine output, and any change in mental status.
Assessment of hemodynamic parameters to identify signs of impending cardiopulmonary collapse (e.g., hypotension, hypoxia, and severe tachycardia) is critical. Physical examination should assess for location of blood loss and signs of systemic illness that may cause anemia. Signs of end-organ dysfunction, such as change in mental status, congestive heart failure, or renal insufficiency should be noted. In the trauma patient, bleeding may be evident or occult, as in the case of femoral, pelvic, or abdominal (including both intra- and retroperitoneal) hemorrhage. These may be hemodynamically significant but not immediately obvious. The presence of trauma itself may be subtle in nonaccidental injury. Look for evidence of gastrointestinal or gynecologic bleeding when the bleeding etiology is unclear.
Management/Diagnostic Testing
Laboratory testing for patients with suspected blood loss includes complete blood count (CBC), reticulocyte count, coagulation studies and a type and screen, or type and cross if transfusion is anticipated. If the etiology of the anemia is unclear, obtain stool guaiac for occult blood, hemolysis labs, and other studies as outlined below (see section on Hemolytic Anemia). Send a pregnancy test if clinically indicated.

FIGURE 101.1 Causes of blood loss.
Management
Initial management steps for all patients include immediate vascular access, cardiorespiratory monitoring, and administration of oxygen, regardless of oxygen saturation to maximize oxygen-carrying capacity of the existing RBC mass and plasma. Unstable patients require multiple sites of vascular access. Peripheral intravenous (IV) catheters are typically more useful than central lines for rapid volume resuscitation; intraosseous access may also be used. If transfusion is anticipated, the blood bank should be notified promptly.
Trauma patients (see Chapter 2 Approach to the Injured Child) require a multidisciplinary team to complete primary, secondary, and tertiary surveys. Occult blood loss as discussed above should be considered in all trauma patients. The use of ultrasound and FAST (focused assessment with sonography in trauma) may be helpful in assessing for intra-abdominal hemorrhage in the multisystem trauma patient (see Chapter 142 Ultrasound). Management is simultaneously directed at immediate hemodynamic stabilization and plans to control blood loss through surgical and catheter-guided embolization strategies as indicated. Patients in uncompensated shock (hypotension, signs of end-organ dysfunction) require rapid fluid resuscitation. Initial crystalloid boluses of 20 ml/kg should be given via pressure bags, rapid infusers, or other means of rapid fluid administration. Patients with uncompensated shock due to hemorrhage despite fluid resuscitation require transfusion (see Table 101.2). While some patients may need immediate transfusion, crystalloid fluids are typically available more quickly and should be used until O-negative blood is available. Hemoglobin changes typically lag behind acute blood loss and are not a useful guide to initial management. Patients with isolated tachycardia but no other signs of end-organ dysfunction such as altered mental status, decreased urine output, or poor perfusion (compensated shock) should be managed based on the severity of the tachycardia, anticipated trajectory of the blood loss, and the need for embolization or operative management. Failure to respond to initial crystalloid resuscitation in these patients may be an indication for transfusion. Traumatic hemorrhage may result in coagulopathy that could worsen bleeding. Trauma patients requiring massive transfusion will need additional blood product support with platelets and fresh-frozen plasma (see Table 101.2) and careful monitoring for electrolyte derangements associated with these treatments. Patients with atraumatic acute blood loss are resuscitated based on the presence of uncompensated shock, on the ability to achieve hemostasis, and the need for procedural intervention. In the stable patient with chronic blood loss, IV fluids should be used with caution, if at all, due to the potential for hemodilution. The decision to transfuse in these cases is based on symptoms, the trajectory of the blood loss, and ability to reverse the inciting cause. Pediatric data are limited, but transfusion in adult patients with hemoglobin >7 g per dL has not improved outcomes. Patients with a hemoglobin below 5 to 6 g per dL frequently require transfusion. In patients with chronic blood loss, evidence of insufficient end-organ perfusion is an indication for transfusion; however, overly rapid or voluminous transfusion can lead to circulatory overload and collapse. For severe anemia with a tenuous hemodynamic status, exchange transfusion may be necessary to safely and efficiently correct the anemia. Specific treatment strategies may be available for some conditions, such as use of estrogen for menorrhagia.
TABLE 101.2
BLOOD PRODUCTS AND DRUGS TO MANAGE BLEEDING



The need for transfusion in a patient with anemia should be carefully considered, given the low but real associated risks of transmission of infectious agents and transfusion reaction. As a practical guide to rapid decision-making, a volume of 10- to 15-ml packed red blood cells (pRBCs) per kg may be given for acute blood loss, with the infusion rate varying from rapid to over 4 hours, depending on the degree of patient instability and rate of ongoing blood loss. In contrast, for severe, chronic anemia, 5-ml pRBCs per kg over 4 hours may be necessary to avoid circulatory overload.
Clinical Indications for Discharge or Admission
In stable patients with anemia, without physiologic compromise, discharge may be considered if the etiology of the condition is known, not expected to progress or accelerate, and close and reliable follow-up is available.
HEMOLYTIC ANEMIA
CLINICAL PEARLS AND PITFALLS
• Promptly evaluate patients with acute onset of pallor and jaundice for hemolytic anemia.
• Autoimmune hemolytic anemia (AIHA) can be life-threatening, especially in cases of severe anemia and reticulocytopenia.
• Early consultation with hematology and the blood bank helps to optimize patient management and to provide sufficient time for compatibility testing if transfusion is needed.
• With evidence of hemolysis, transfusion indications include symptomatic anemia or low presenting hemoglobin (<5 to 6 g per dL in children, <6 to 7 g per dL in adolescents).
• Avoid excessive crystalloid fluid resuscitation in severely anemic patients with hemolytic process.
Current Evidence
The premature destruction of RBCs in circulation (hemolytic anemia) is most commonly due to intrinsic defects of erythrocytes (membranopathies, enzymopathies, or hemoglobinopathies) in pediatric patients; however, extrinsic or extracorpuscular factors such as antibodies, environmental stresses, infection, or microangiopathic damage may also cause hemolysis. The severity of anemia can range from mild to life-threatening and is often influenced by the underlying mechanism. For example, erythrocyte membrane disorders (hereditary spherocytosis, hereditary elliptocytosis, hereditary stomatocytosis) and metabolic abnormalities (glucose-6-phosphate dehydrogenase [G6PD] deficiency, pyruvate kinase deficiency) do not usually cause severe anemia and rarely constitute a life-threatening emergency. AIHA, on the other hand, may present with a precipitously falling hemoglobin level in a child who appears critically ill with signs of congestive heart failure. Reported mortality rates for pediatric patients with hemolytic anemia range from 4% to 10%. Fortunately, this is a rare condition in children.
Goals of Treatment
Successful treatment of hemolytic anemia requires stabilization of the hemoglobin level and maintenance of sufficient oxygen-carrying capacity and cardiac output. Clinical outcomes depend on early recognition and intervention for uncompensated anemia.
Clinical Considerations
Clinical Recognition
Hemolytic anemia arises in all age groups. The typical presenting features include pallor, jaundice, and dark urine. Other symptoms include fatigue, malaise, dizziness, fever, and abdominal or back pain. Symptoms often develop suddenly, and in cases of rapid hemolysis, the presenting hemoglobin level may be as low as 3 to 4 g per dL. Some patients follow an indolent course presenting with symptoms evolving over days or weeks. Any unexplained low hemoglobin level, typically accompanied by a reticulocytosis, should prompt consideration of a hemolytic anemia.
Triage
Children with hemolytic anemia often present in a relatively compensated state; however, their clinical status can deteriorate rapidly. Severe, uncontrolled hemolytic anemia can be fatal with patients succumbing to insufficient oxygen-carrying capacity and cardiovascular collapse. Ongoing assessment of cardiovascular and neurologic status is crucial during the diagnostic evaluation and until the rate of hemolysis is controlled. Alert the hematology service and blood bank to patients with suspected hemolytic anemia early in the evaluation process.
Initial Assessment/H&P
The history should focus on systemic complaints relevant to anemia and hemolysis such as fatigue, light-headedness or near-syncope, fussiness/irritability in young children, and dyspnea. Probe for symptoms of a systemic autoimmune process, an immunologic disorder, and malignancy as these can be associated with AIHA. Elicit any personal or family history of an intrinsic RBC defect or AIHA. Attention to recent exposures including symptoms suggestive of a recent or concurrent viral illness, toxins (e.g., naphthalene-containing mothballs), and medications can also provide clues about possible triggers (Table 101.3). Typical examination findings for patients with hemolytic anemia include jaundice/icterus, pallor, tachycardia in some cases accompanied by murmur from a high output cardiac state, and mild hepatosplenomegaly. Other findings such as lymphadenopathy or massive hepatosplenomegaly may suggest underlying infection or malignancy. The presence of jugular venous distention, significant hepatosplenomegaly, gallop rhythm, respiratory distress, hypotension, or poor perfusion heralds imminent cardiovascular collapse.
Diagnostic Testing
A basic approach to the evaluation of hemolytic anemia is presented in Figure 101.2. The initial laboratory evaluation should include a CBC and differential, peripheral blood smear, reticulocyte count, blood type and antibody screen (indirect antibody test, formerly known as an indirect Coombs test), direct antiglobulin test (DAT, formerly called a direct Coombs test), haptoglobin, serum electrolytes, BUN/Cr, and bilirubin level. The goal is to identify a hemolytic anemia, prepare for transfusion in case the hemoglobin is critically low or falling rapidly, and narrow the differential of the causative mechanism. Reticulocytosis is present in most cases but can be absent or delayed in up to 10% of patients. Consideration of hemolytic anemia should not be eliminated based solely on a low reticulocyte count. Distinguishing between an immune- and a non–immune-mediated hemolytic anemia is an essential first step as this dictates therapy. The DAT uses broad-spectrum Coombs serum (IgG, IgM, and complement) to detect the presence of antibodies on erythrocytes and is usually positive in AIHA. A positive DAT prompts additional testing by the blood bank to further characterize the antibody. Acute hemolysis is most commonly associated with a warm (37°C)-reactive IgG antibody with or without complement (C3). This type of hemolysis is usually extravascular, occurring in the spleen. IgM-mediated cold agglutinin disease is less common in children than in adults, but can occur following mycoplasma (anti-I) or infectious mononucleosis (anti-i). These antibodies bind to RBCs in the cold and characteristically cause intravascular hemolysis as complement is bound and activated at warmer temperatures. Cold-reactive IgG antibodies (Donath–Landsteiner test) cause paroxysmal cold hemoglobinuria (PCH), which in children frequently follows a viral infection. A negative DAT does not definitively exclude an immune-mediated process. Rarely, IgA or warm reactive-IgM antibodies may be present but not detected by the Coombs reagent. Additionally, in rare cases, the causative warm-IgG antibodies are below the level of detection. When there is a high degree of suspicion for an immune-mediated process despite a negative DAT, specialized assays are required for antibody detection.
TABLE 101.3
EXTRACORPUSCULAR CAUSES OF HEMOLYTIC ANEMIA


Examination of the peripheral blood smear is helpful in diagnosing subsets of nonimmune hemolytic anemia especially in the acute care setting when the results of disease-specific testing such as osmotic fragility, hemoglobin electrophoresis, or G6PD activity will not be available. Certain patterns of red cell morphology can support a diagnosis of hemoglobinopathy or enzymopathy. The presence of RBC fragments (schistocytes) on the peripheral smear suggests mechanical damage to the erythrocyte. If the diagnosis is uncertain, a pretransfusion blood sample should be saved for additional testing such as the measurement of specific enzyme levels or hemoglobin electrophoresis. The diagnosis of microangiopathic processes such as hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP) is important and should be considered in patients with diarrhea, and in the presence of acute kidney injury, thrombocytopenia, and fever and neurologic changes. Disseminated intravascular coagulation (DIC) occurs in the context of systemic illness, including sepsis, trauma, or malignancy. If considering any microangiopathic process, evaluation should also include a coagulation panel.
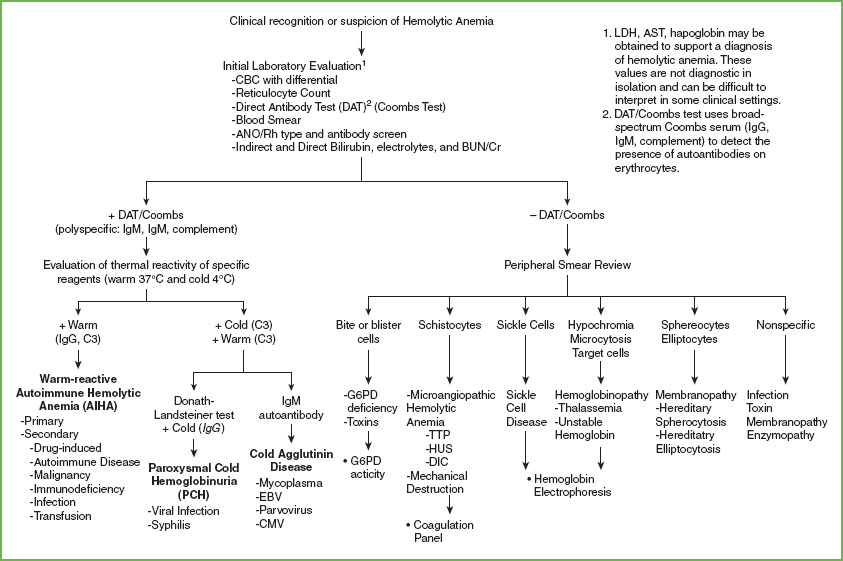
FIGURE 101.2 Approach to evaluation of hemolytic anemia.
Management
AIHA. Prompt initiation of therapy for AIHA is necessary to stabilize the rate of hemolysis. Hospitalization for careful observation and treatment is usually necessary; the hemoglobin level may continue to fall precipitously despite initial therapy. Patients with symptomatic or severe anemia (hemoglobin <5 to 6 g per dL in children, <6 to 7 g per dL in adolescents) should receive a pRBC transfusion. Hemolytic transfusion reactions are a potential hazard. Transfuse an initial 5 to 10 mL of blood slowly to test for a significant hemolytic transfusion reaction associated with the selected aliquot of blood. If the patient does not manifest signs or symptoms of acute worsening during the initial infusion, increase the transfusion rate. Ultimately, the transfusion rate must exceed the rate of ongoing hemolysis. Careful monitoring of hemoglobin levels is important, with a stable hemoglobin goal of 6–8 g per dL.
The management strategy differs for those with a warm-reactive antibody versus those with a cold-reactive antibody as highlighted in Figure 101.3. Hemolysis and erythrocyte clearance in the setting of a warm-reactive antibody is generally extravascular, taking place in the spleen. These patients typically respond to corticosteroid therapy, but may also benefit from concomitant IVIG or splenectomy in more dire circumstances. Cold-reactive AIHA is intravascular hemolysis, dominated by complement activation and with limited response to corticosteroids or splenectomy. These patients should be kept warm (i.e., avoid cold exposure), and they may benefit from plasmapheresis in severe cases. In the setting of intravascular hemolysis, accumulation of circulating free hemoglobin and cellular contents can be toxic to the renal tubular system. Careful fluid management to ensure adequate renal clearance without excessive dilution of the red cells is imperative.
Nonimmune Hemolytic Anemia. Most etiologies of nonimmune hemolytic anemia require observation and supportive care, including removal of the offending agent and prevention of renal damage due to significant hemolysis (see Table 101.3). Infectious agents that may induce hemolytic anemia include malaria, other protozoa, and a wide variety of gram-positive and gram-negative organisms that require identification and prompt treatment. When hemolysis is a result of small vessel disease, treatment of the underlying disorder (e.g., collagen vascular disease) or primarily affected organs (e.g., renal failure in hemolytic-uremic syndrome) is the first priority. The prompt institution of plasma exchange for TTP can be lifesaving.
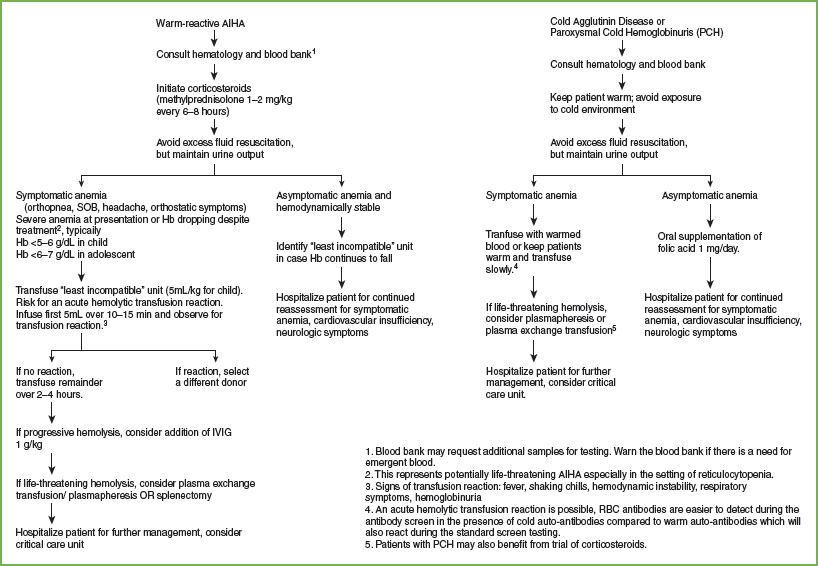
FIGURE 101.3 Management of autoimmune hemolytic anemia.
Clinical Indications for Discharge or Admission
Hospitalize patients with severe or symptomatic anemia or an unclear clinical trajectory for close clinical monitoring and treatment. Frequently a critical care setting is appropriate for these patients. Consider outpatient management in patients with a clear or well-established underlying diagnosis and mild anemia with short interval follow-up for monitoring and ongoing management.
METHEMOGLOBINEMIA
Goals of Treatment
Methemoglobin (MHb) is the end product of a number of mechanisms (toxic exposure, dietary trigger, acidosis, genetic abnormality) that oxidize the iron associated with a heme group from the ferrous (Fe2+) to ferric state (Fe3+) rendering it unable to reversibly bind oxygen. In high quantities, insufficient gas exchange may be incompatible with life. The primary goal of treatment is to remove the causative agent, provide supportive care to optimize end-organ oxygenation, and allow time for reduction of MHb back to hemoglobin. In symptomatic and life-threatening situations, therapeutic intervention can hasten the reduction process.
CLINICAL PEARLS AND PITFALLS
• Suspect MHb when a cyanotic patient has a normal arterial PO2, and pulse oximetry (generally in the mid-80s) is significantly lower than the oxygen saturation reported on arterial blood gas.
• MHb levels are reported as percent of total hemoglobin; therefore, patients with anemia will manifest more symptoms at lower levels of MHb.
• Methylene blue administration for methemoglobinemia is contraindicated in patients with G6PD.
Clinical Considerations
Methemoglobinemia is an uncommon cause of cyanosis in infants and children, but can cause significant morbidity and even death. Oxidant stress under physiologic conditions produces MHb that is reduced back to hemoglobin by cellular mechanisms. In normal individuals, MHb exists in a steady state of about 1% of total hemoglobin; this may be higher in those chronically exposed to tobacco smoke. Congenital forms of methemoglobinemia present in the neonatal period or early infancy and result from defects in or absence of the endogenous reductase systems. Defects in globin chains (α- or β-chain) can also alter the oxidation state of the heme iron resulting in cyanosis. Termed M hemoglobins, they are inherited in an autosomal dominant fashion and typically do not require treatment. Acquired forms of methemoglobinemia are more common. Ingestion or topical exposure to oxidizing drugs or chemicals (benzocaine, dapsone, chloroquine, nitrates, paraquat) occurs most commonly. Ingestion of high levels of nitrates, such as through well water, can cause MHb. Systemic acidosis in infants may also result in MHb because of the relative immaturity of NADH-dependent enzyme system early in life.
TABLE 101.4
SYMPTOMS ASSOCIATED WITH MHb LEVELS
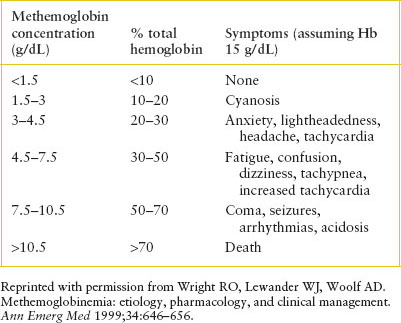
Clinical Recognition
Consider the diagnosis of methemoglobinemia when cyanosis occurs in the absence of cardiac or pulmonary disease and does not improve with oxygen therapy. Symptoms depend on the actual concentration of MHb (see Table 101.4). At low levels, patients may present with cyanosis only. As the level rises, symptoms of headache, fatigue, anxiety, and lightheadedness develop. A MHb level greater than 30% is considered life-threatening, and these patients may exhibit altered mental status, dyspnea, tachypnea, tachycardia, seizures, respiratory depression, and arrhythmia. Coma may occur at MHb levels above 50%.
Management/Diagnostic Testing
The treatment of methemoglobinemia depends on the clinical severity. In all cases, attempt to identify and remove the causative oxidant stress. If symptoms are mild after oxidant exposure, therapy may be unnecessary. RBCs with normal metabolism will reduce the MHb in several hours. In general, treat patients with MHb level >20% with 1 to 2 mg per kg of methylene blue as a 1% solution in saline infused intravenously over 5 minutes. Administer a second dose if symptoms are still present 1 hour later. Patients with a significant concurrent medical condition, especially cardiopulmonary conditions, should be considered for treatment at MHb levels starting at 10%. Methylene blue is an oxidant at high dosages, so total dosage should not exceed 7 mg per kg to avoid paradoxical methemoglobinemia.
Use methylene blue with extreme caution in patients with G6PD due to the risk of hemolytic anemia. The mechanism of action of methylene blue relies on NADPH. These patients may not produce sufficient quantities of NADPH to respond to this therapy; however, some patients have partial enzyme activity. Methylene blue at lower doses (0.3 to 0.5 mg/kg/dose) may lower MHb levels without causing significant hemolytic anemia. The addition of ascorbic acid 5 to 8 mg/kg/day may benefit G6PD patients. Consider exchange transfusion in patients who fail methylene blue treatment or have absent G6PD activity.
Clinical Indications for Discharge or Admission
Even if treatment with methylene blue or ascorbic acid in the ED is successful, admit any child with symptomatic methemoglobinemia to the hospital for close observation, especially if the etiology is unknown. Some oxidizing agents such as dapsone and aniline have been reported to cause cyclic or rebound methemoglobinemia. Consultation with toxicology or poison control is advised.
INEFFECTIVE OR DECREASED RED BLOOD CELL PRODUCTION
CLINICAL PEARLS AND PITFALLS
• Decreased RBC production may be due to congenital abnormalities (Diamond–Blackfan anemia [DBA]), marrow infiltrative processes (malignancy, fibrosis, lysosomal storage disease), or acquired insults (viral suppression, toxins, nutritional deficiency).
• β-Thalassemia major usually presents only after 2 to 3 months of age with the switch from γ- to β-globin chain production; it may present with severe anemia causing a hematologic emergency.
• Iron replacement therapy for iron-deficiency anemia consists of 3 to 6 mg/kg/day of elemental iron given orally as ferrous sulfate at night on an empty stomach (and ideally with ascorbic acid) as a single daily dose. IV or SQ formulations do not provide an advantage in the absence of malabsorption.
• The goal of transfusion for chronic, severe anemia is relief of symptoms, not restoration of a normal hemoglobin level. If transfusion is necessary, provide small aliquots of RBCs (5 mL per kg) slowly with close monitoring and consideration of furosemide administration.
Current Evidence
Thalassemia
The thalassemias are mutations or deletions of the globin genes that result in reduced or loss of production of α- or β-globin chains, the building blocks of hemoglobin. Hemoglobin A (normal adult hemoglobin α2β2) begins rising just prior to birth as the body switches production from γ- to β-globin chains, and increases steadily over the first year of life to become the predominant hemoglobin.
The β-thalassemia gene mutations occur most commonly in populations living in regions bordering the Mediterranean, North Africa, the Middle East, Central and Southeast Asia, and India. In β-thalassemia, reduced (β+) or no (β0) β-globin chains are produced. β-Thalassemia major phenotype results from β0/β0 or β0/β+, intermedia phenotype from β+/β+, and carrier state from β/β+.
In the first year of life, infants with β-thalassemia major usually develop a sallow complexion, increased fatigue, and poor weight gain and linear growth. Physical examination shows pallor, icterus, and enlargement of the liver and spleen. In thalassemia major, the hemoglobin level may decrease to 3 or 4 g per dL, and the mean corpuscular volume is low. The RBCs are hypochromic and microcytic, with striking variation in size and shape; nucleated RBCs are present on the peripheral smear. Children and adolescents with thalassemia intermedia have a moderate anemia, with hemoglobin levels usually between 7 and 10 g per dL. Hemoglobin electrophoresis is used to make the initial diagnosis of β-thalassemia, although in many cases index of suspicion is high due to family history. Hemoglobin electrophoresis may also reveal other mutant β-chains such as hemoglobin E or C. Independently, these hemoglobins may not cause a significant clinical phenotype; however, in combination with a β0 or β+ mutation, patients may have a β-thalassemia major or intermedia phenotype.
Patients with β-thalassemia major require lifelong transfusion or bone marrow transplantation. They may present to the emergency department with symptoms of severe anemia prior to initial diagnosis or later in life due to toxicity related to iron overload (often due to poor compliance with iron chelation therapy). The β-thalassemia intermedia phenotype is variable, but patients typically only need transfusions for acute exacerbations of their anemia during illness, pregnancy, or perioperatively. Patients who carry one-mutated β-gene are asymptomatic but their red cells are microcytic (low MCV) and a mild anemia may be evident.
The α-thalassemia gene mutations occur most commonly in populations living in Mediterranean countries, northern Africa, the Middle East, India, and Southeast Asia. Loss of one or two of the four α-globin genes is clinically trivial and manifests as a silent carrier or α-thalassemia trait, respectively. Loss of three α-globin genes causes hemoglobin H disease, usually associated with a moderate anemia and chronic hemolysis. Loss of all four α-globin genes results in production of Hemoglobin Barts (γ4) and hydrops fetalis (stillborn or death soon after birth). Hemoglobin electrophoresis does not aid the diagnosis of α-thalassemia trait but can identify Hemoglobin H. Hemoglobin Barts can be detected on newborn screen.
Congenital and Acquired Aplastic/Hypoplastic Anemia
The differential diagnosis of aplastic and hypoplastic anemias is discussed in Chapter 57 Pallor. DBA and transient erythroblastopenia of childhood (TEC) are the more common causes of pure RBC aplasia in early childhood. In DBA, the level of RBC adenosine deaminase (ADA) is frequently elevated, and blood samples for this test should be obtained before transfusion. TEC typically presents in previously healthy children from infancy to toddlerhood with severe anemia and reticulocytopenia but otherwise normal blood counts. For patients with a hypoplastic anemia suggestive of TEC, a bone marrow aspirate may be helpful in predicting the course of the disease during the next few days and, in particular, the likelihood that pRBC transfusions will be required. For example, if a patient with TEC has a hemoglobin level of 4 g per dL, low reticulocyte count, and few RBC precursors on bone marrow evaluation, a further decrease in the hemoglobin concentration should be anticipated and pRBC transfusions will almost certainly be required. However, if the bone marrow aspirate shows numerous erythrocyte precursors progressing through all levels of erythrocyte maturation, a peripheral reticulocytosis can be expected within 24 hours and RBC transfusions may be unnecessary.
Nutrition Deficiencies and Excess
Nutritional anemias in children constitute more of a public health problem than a hematologic emergency; however, in some cases the hemoglobin level may be very low at the time of diagnosis. Some of the most common micro-nutrients related to anemia include deficiencies of iron, B12, folate, zinc, vitamin C, and excess of copper. Of these, iron-deficiency anemia is the most common in the pediatric population.
Severe iron deficiency occurs mainly in toddlers due to excess cow’s milk consumption (more than 1 quart [32 fl oz or ∼1 L] daily). Adolescent girls make up another group at high risk for iron deficiency because a diet normally marginal in iron content becomes inadequate in the face of menstrual blood losses. The presenting complaints in severe iron-deficiency anemia include pallor, lethargy, irritability, or poor exercise tolerance. Iron replacement therapy consists of 3 to 6 mg/kg/day of elemental iron given orally as ferrous sulfate at night on an empty stomach (and ideally with ascorbic acid) as a single daily dose. The hematologic response to parenterally administered iron is no faster than the response to orally administered iron for patients with intact gastrointestinal absorption. Historically, intravenously administered iron has been associated with anaphylaxis, but such reactions are rare with modern preparations.
Vitamin B12 and folate deficiency result in megaloblastic macrocytic anemia. Infants exclusively breast-fed by a vegetarian mother may develop vitamin B12 deficiency. In folic acid deficiency caused by impaired folate absorption, nonhematologic symptoms such as diarrhea, slowed development, or altered mental status and coma may be more prominent than the symptoms of anemia. When considering replacement of folic acid or vitamin B12, traditional replacement doses of 1 mg of folic acid and 100 mcg of vitamin B12 daily are undoubtedly excessive, but their common use reflects the safety and concentrations of the available compounds. The administration of supplemental iron, vitamin B12, or folic acid should not be considered a substitute for adequate dietary intake when nutritional deficiency is recognized. Unlike most hematologic emergencies, the rapid improvement after treatment of these disorders may reduce the likelihood of further visits despite attempts to ensure adequate follow-up care. Therefore, a strong effort to restructure the diet should begin at the time of the initial contact.
Clinical Considerations
Clinical Recognition
Patients with congenital and acquired disorders of effective RBC production may be detected incidentally, with mild symptoms such as pallor, fatigue or malaise, or due to severe symptoms such as cardiopulmonary compromise. Some viral infections, such as parvovirus, may precipitate an anemic crisis in a previously stable patient with a chronic process due to suppression of erythropoiesis. A mild anemia due to chronic disease may be the presentation of a child with systemic illness. Age of onset helps to guide differential diagnosis. While acquired etiologies can arise at any age, congenital forms of aplastic anemia and thalassemia major typically present during infancy or early childhood.
Initial Assessment/H&P
History should include a thorough review of systems including general complaints of fatigue or decreased exercised tolerance, dietary history, family history and ethnic origin, and menstrual history where appropriate. Physical examination should assess for lymphadenopathy, organomegaly, and signs of bleeding.
Diagnostic Testing
Laboratory testing should include a CBC, reticulocyte count, review of peripheral blood smear, type and cross, reticulocyte hemoglobin (CHr), indirect antibody test (i.e., indirect Coombs test), and DAT (i.e., Coombs test), total and direct bilirubin, ferritin, plasma iron, total iron binding capacity (TIBC), chemistry panel, urinalysis, and a stool examination for occult blood. Additional testing including hemoglobin electrophoresis, micronutrient levels, ADA activity, genetic testing for congenital aplastic anemias may also be appropriate and should be guided by consultation with a hematologist. Often these subspecialized tests are not interpretable in the weeks following a transfusion, so communication with hematology prior to transfusion is preferable if clinical situation permits.
Management
The decision regarding transfusion of pRBCs depends on the etiology and severity of the anemia. Replacement of nutrient deficiencies and elimination of toxin exposures is necessary (see Table 101.5). If the hemoglobin level is mildly decreased from baseline levels and the patient has no evidence of cardiovascular compromise, transfusion may be unnecessary. However, transfusion should be considered in any patient with cardiovascular compromise, or borderline cardiovascular symptoms in the setting of expected ongoing losses. The goal of transfusion should be relief of symptoms, not restoration of a normal hemoglobin level. If transfusion is necessary, provide small aliquots of pRBCs (5 mL per kg) slowly. The administration of a rapid-acting diuretic (furosemide 1 mg/kg/dose, maximum 20 mg per dose) may diminish the risk of fluid overload.
For patients requiring recurrent or chronic transfusions or patients who may be candidates for bone marrow transplantation, blood bank measures to provide CMV-negative products and reduce the likelihood of alloimmunization by leukoreduction of pRBC units or extended RBC antigen typing are beneficial. First-degree relatives should not be chosen as blood donors to avoid allosensitization to family minor HLA antigens.
SICKLE CELL DISEASE
Goals of Treatment
Patients with sickle cell disorders suffer from three broad categories of complications: vascular occlusion, infection, and end-organ damage. Immediate recognition and early management of these complications can prevent hospitalizations, decrease the need for significant interventions such as exchange transfusions, and avoid long-term morbidity and death.
CLINICAL PEARLS AND PITFALLS
• Sickle cell patients require aggressive pain control when they present with a vasoocclusive episode. Management includes IV fluids to improve vascular flow, nonsteroidal anti-inflammatory drugs (NSAIDs), and administration of narcotics.
• Acute chest syndrome presents as fever, respiratory symptoms (increased work of breathing, hypoxia, or chest pain), and a new infiltrate on CXR. Acute chest syndrome can evolve rapidly to become life-threatening.
• Patients with sickle cell disease have a 300-fold higher risk of stroke compared with other children. New neurologic findings should prompt emergent hematology consultation, plans for exchange transfusion, and head imaging.
• Patients are functionally asplenic and at increased risk of severe bacterial infections. Fever >101.5°F/38.5°C necessitates blood culture (other cultures as clinically indicated) and empiric coverage with a broad-spectrum antibiotic such as ceftriaxone.
• Splenic sequestration results from pooling of blood within the spleen and can lead to hypovolemic shock necessitating transfusion. Most presentations are in children <3 years old; however, later presentations can occur in patients with HbSC or Hb Sickle β-thalassemia.
TABLE 101.5
TREATMENT OF ANEMIA

Clinical Considerations
Clinical Recognition
Patients with sickle cell disease require prompt evaluation for complications including stroke, vasoocclusive episodes, acute chest syndrome, splenic sequestration, infection, and priapism. Consider the diagnosis of sickle cell disease in genetically susceptible children with unexplained pain or swelling (especially of the hands or feet), pneumonia, meningitis, sepsis, neurologic abnormalities, splenomegaly, or anemia. Recognition of sickle cell disease is key to appropriate intervention. Definitive testing to diagnose sickling disorders, hemoglobin electrophoresis, is not usually performed in the ED.
Triage
Immediately evaluate any sickle cell patient with fever, concern for stroke, acute chest syndrome, splenic sequestration, or priapism.
Vasoocclusive Pain Episodes
Initial Assessment/H&P
Vasoocclusive pain episodes are responsible for nearly 90% of all emergency department visits and 70% of hospitalizations related to sickle cell disease. Pain may be severe and debilitating for patients. Many patients live their entire lives with some degree of baseline pain. Cold exposure and dehydration trigger increased sickling. Joint pain presents a diagnostic challenge because vasoocclusive pain and the symptoms of trauma and infection (septic arthritis and osteomyelitis) present similarly and the physical examination findings, laboratory tests, and imaging features are often nonspecific. For a given patient, pain that is typical and/or at a recurrent location supports a vasoocclusive etiology. Dactylitis (infarction of the metacarpals, metatarsals, and phalanges) usually results in swelling of the hands and feet and is a common presentation in children 6 months to 2 years of age. Pain usually resolves after several days, but swelling may persist for 1 to 2 weeks.
Management/Diagnostic Testing
No definitive test exists for diagnosing a vasoocclusive episode. Hydration and prompt pain management are key (see Table 101.6
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


