Chapter 71 Glomerulotubular Dysfunction and Acute Kidney Injury
Acute kidney injury (AKI) is a frequent problem in the pediatric intensive care unit (PICU), but an accurate incidence is difficult to establish due to the ongoing evolution of a clear definition of renal failure. The classic definition was a 50% reduction of glomerular filtration rate (GFR) accompanied by a 50% increase in creatinine. A RIFLE classification of renal injury to allow earlier appreciation of renal dysfunction is now utilized in adults and a modification of this system (pRIFLE) can also be applied to children.1–3
Early detection of renal dysfunction by biomarkers is needed, to allow a more timely awareness of injury and, thereby, modifications to alleviate the renal stress. Recent investigations in this area, although early in their development, postulate a panel approach (plasma panel of neutrophil gelatinase-associated lipocalin [NGAL] and cystatin C or a urine panel of NGAL, interleukin 18, and kidney injury molecule-1 [KIM-1]) may be of help in the future to provide more sensitive and specific detection of early AKI.4–6
Acute Kidney Injury Pathophysiology
Physiology of Glomerular Filtration
GFR is the product of the filtration rate of the individual nephron and the number of functioning nephrons.7 A single nephron glomerular filtration rate (SNGFR), is defined by the Starling forces of the glomerular capillaries and the properties of the glomerular capillary wall.
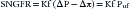
where Kf is a capillary wall property known as the ultrafiltration coefficient and is the product of the surface area available for filtration and the hydraulic conductivity of the membrane. The Starling forces (or pressures) that affect filtration are the hydraulic pressure in the glomerular capillary (Pgc), the hydraulic pressure in Bowman’s space (Pbs), the oncotic pressure of the glomerular capillary (πgc), and the oncotic pressure in Bowman’s space (πbs), which is usually zero because the ultrafiltrate is essentially protein-free.8 Pgc favors filtration; Pbs and πgc are opposing forces to filtration. The mean ultrafiltration pressure (Puf) is the difference between the net change in hydraulic pressure and the net change in the oncotic pressure. Thus SNGFR may be modified by alterations in the glomerular capillary pressures, glomerular membrane characteristics, or the surface area available for filtration.
Glomerular filtration depends on adequate renal perfusion; the kidneys receive approximately 25% of total cardiac output. The fraction of cardiac output perfusing the kidneys is related to the ratio of renal vascular resistance (RVR) and systemic vascular resistance.9 Renal blood flow (RBF) is determined by systemic blood pressure (SBP) and RVR, expressed by the formula RBF = SBP/RVR.9 Kidney autoregulation, which maintains a constant renal perfusion pressure, occurs through alterations in RVR in response to changes in systemic vascular resistance or intravascular volume. When SBP is within the normal physiological or autoregulatory range, the kidney can maintain constant blood flow and GFR by dilation of the preglomerular or afferent arteriole, which reduces RVR and increases RBF. This afferent arteriole dilation is accomplished by two known mechanisms, smooth muscle relaxation of the afferent arteriole in response to sensing a transmural pressure drop (the myogenic reflex3) and the tubuloglomerular feedback system. The tubuloglomerular feedback system is operational following a reduction of plasma flow. When solute and water delivery to the macula densa are reduced, the juxtaglomerular apparatus responds by relaxing the smooth muscle of the adjacent afferent arteriole. Thus a reduction in cardiac output or effective renal plasma flow is accompanied by vasodilation at the preglomerular arteriole, which in turn reduces RVR, thereby restoring RBF.
During states of reduced cardiac output or intravascular volume depletion, the systemic vasoconstrictors, angiotensin II and vasopressin, are released to help preserve vascular tone. The kidney counteracts the renal vasoconstrictor activity of angiotensin II and increased sympathetic tone through the intrarenal production of vasodilatory prostaglandins such as prostaglandin I2.10 These locally produced substances may attenuate renal vasoconstrictive forces and help preserve renal perfusion. Animal model data of congestive heart failure have provided evidence that enhanced prostaglandin synthesis is required for preservation of renal perfusion and GFR. Patients receiving prostaglandin synthetase inhibitors such as nonsteroidal antiinflammatory drugs (NSAIDs) have potentiation of renal ischemia because of an increase in renal vasoconstriction not antagonized by intrarenal prostaglandin synthesis.11 Endothelium-derived relaxation factors (EDRFs), which are vasodilatory, and the potent vasoconstrictor endothelin, produced by the endothelium, may also affect the regional vascular tone.12
Constriction of the postglomerular capillary sphincter, the efferent arteriole, in the face of reduced RBF serves to increase the filtration fraction and preserve GFR, although this occurs at the expense of renal plasma flow, which may be further reduced. Vasoconstriction at the efferent arteriole is mediated by angiotensin II and, to a lesser extent, by the action of the adrenergic system by epinephrine.13 Elevation in postglomerular arteriolar resistance may be blocked by the angiotensin-converting enzyme inhibitors. When converting enzyme inhibitors are administered to the patient who requires efferent arteriolar constriction to maintain GFR, renal decompensation often results.14
As previously noted, reductions in effective intravascular volume and cardiac output are accompanied by increased activity of the sympathetic nervous system and the renin-angiotensin-aldosterone system and increased circulating levels of vasopressin.15 Hormonal and neural systems signal the kidneys to increase the reabsorption of sodium and water to help restore the deficient intravascular volume, increase cardiac output, and consequently improve RBF. These, and the kidney’s homeostatic mechanism, by afferent arteriolar vasodilation and efferent arteriolar constriction, maintain the kidney’s glomerular filtration. The kidney’s homeostatic mechanisms, however, are not without limitation. The autoregulatory ability of the afferent arteriole is maximal once the mean SBP falls below 80 mm Hg. The renal autoregulatory range appears to be age-dependent, because younger animals can autoregulate over lower pressure ranges. The range of perfusion pressure over which the kidney can autoregulate may be limited in certain conditions so that vasodilation is maximal with a minor reduction in mean arterial blood pressure. Examples include extracellular fluid depletion, renal ischemia, or renal vascular disease (e.g., hypertension, diabetes, atherosclerosis).
Pharmacological agents may alter renal perfusion by changing SBP through an action on systemic vasculature or by direct effects on renal vasculature (Box 71-1). Vasodilators, such as hydralazine, lower SBP without changing renal perfusion pressure, because the decrease in SBP is accompanied by decreased RVR. Conversely, epinephrine increases SBP but decreases RBF by its vasoconstrictor effect on intrarenal blood vessels.
Box 71–1 Vasoactive Substances in the Kidney Vasculature
Data from Hostetter TH, Brenner BM: Kidney circulatory and nephron function in experimental acute kidney failure. In Brenner BM, Lazarus JM, editors: Acute kidney failure, ed 2, New York, 1988, Churchill Livingstone.
Morphologic Changes in Renal Injury
The kidney’s complex structure, with heterogeneous segments within the kidney receiving differential regional perfusion and thereby oxygenation, sets up a common form of renal failure, that of the tubulointerstitium. This region is at greatest risk for ischemia because of its gradient of regional perfusion and oxygenation. In addition, vascular disease, including glomerular disease, often occurs in children and results in AKI, but there are studies that suggest the extent of damage to the tubulointerstitium has the greatest prognostic implication to the degree of final renal recovery.16
Initial structural changes in tubular cells are seen as apical and basal surface changes of simplification, with microvilli of the brush border shortening and disappearing by either detachment from the apical surface or being internalized within the tubular cells.17
In this scenario, enzymes of the brush border (alkaline phosphatase and gammaglutamyl transpeptidase), may be found in the urine and may be used as markers of early tubular injury.18 The loss of microvilli surface area leads to loss of enzymes and transport sites for transcellular absorption and apical uptake. Additionally, loss at the basolateral interdigitating infoldings of the tubular cells then results in further reduction of surface area for transport and loss of the Na-K-adenosine triphosphatase (ATPase) that is localized to this membrane and involved in many transport processes.19
Morphologic changes in distal tubules and outer medulla also occur and may be found even when proximal tubular injury is not readily identified on biopsy. Experimentally, the outer medulla has been identified as sensitive to hypoxia, including that induced by toxins such as radiocontrast agents and cyclosporine.20
Injury at this outer medulla region may be missed on biopsy since this site often is not sampled. Tubular cell detachment with exposed, denuded regions of the basement membrane can be found on biopsies, as a result of altered cell-matrix attachments.21
Renal tubular sensitivity to ischemic injury is primarily influenced by the individual renal cell’s energy requirements, its glycolytic capacity, and the extent of hypoxic stress upon the cell. Glycolysis and oxidative phosphorylation both supply the ATP required by the cell to drive its metabolism, and it follows that cells with a greater capacity for glycolysis (distal tubular cells) are less sensitive to oxygen deprivation than the cells that rely mainly on mitochondrial derived energy, (proximal tubular cells).22
In vivo and in vitro studies may be discordant in identifying susceptiblity to hypoxia. Medullary straight portions of the proximal tubule have a higher glycolytic capacity than the cortical segments of the proximal tubule but, due to the regional distribution of perfusion within the kidney, the medullary portions operate in a lower oxygen-tension environment and therefore are more susceptible to hypoxia/ischemic injury.23 The individual cell’s energy requirements to conduct its transport activities further influences its risk to hypoxic injury. The outer medullary proximal tubular cell (pars recta), due to the high energy requirements for its transport functions, has a greater injury risk than the deep inner medullary tubular cell with its low-oxygen environment, but also a low energy requirement to maintain its transport function.24
When energy stores are rapidly depleted, the normal Na-K ATPase pump begins to fail and the most basic of cell function, membrane integrity, is jeopardized, with resultant accumulation of Na, Cl, and water within the cell; this cell swelling (oncosis) is a hallmark feature of necrosis.25 Not all cells that fail to maintain their energy requirements die through necrosis; apoptosis, which appears morphologically as cell shrinkage, is also a result of inadequate energy support. Apoptosis is an asynchronous cell death triggered over hours to days, with the earliest cellular element involving mitochondrial changes including loss of transmembrane potential and release of mitochondrial cytochrome c into the cytosol.30 It is mitochondrial function/dysfunction that primarily determines the fate of the cell for recovery and survival or death, and by what form: apoptosis or necrosis.
Pathogenesis of Reduced Glomerular Filtration Rate in Acute Kidney Injury
The mechanisms responsible for GFR reduction in acute renal injury have been studied extensively with experimental models of AKI. Multiple mechanisms are often operational in mediating hypofiltration. Whereas one factor may have greater importance in the initiation of injury and decreased filtration, others are involved in the sustained reduction in GFR during the maintenance phase of AKI. Four major mechanisms result in reduced GFR during AKI: reduced blood flow, decreased Kf, tubular obstruction, and backleakage of tubular fluid.31 Each factor is discussed regarding its role in both the initiation and maintenance phases of AKI.
A reduction in RBF can be demonstrated during the initiation phase of many forms of AKI and seems to play a predominant role in ischemic injury and rhabdomyolysis.31 Proposed theories for the reduction of RBF include (1) a proportional increase in the afferent and efferent arteriolar resistances in response to activation of the renin-angiotensin system; (2) vascular endothelial cell swelling and damage with release of vasoactive peptides such as endothelin; and (3) hyperemic congestion of the medullary peritubular capillaries.
Intratubular obstruction occurs in most forms of acute renal injury, either as a contributing factor in the initiation phase or during the maintenance phase.31 Tubular obstruction with cellular debris and precipitated protein is a prominent finding in both the initiation and maintenance phases of ischemic injury. In the case of nephrotoxic injury, the degree of injury may determine the extent of tubular obstruction. In an experimental model of gentamicin nephrotoxicity, the drug dose was positively correlated with the contribution of tubular obstruction to reduced GFR.31 Tubular obstruction and loss of epithelial integrity caused by tubular cell injury result in the backleakage of tubular fluid and solutes. Excretion of solute and fluid is decreased, and this decrease possibly signals a further reduction in the GFR by stimulation of the tubuloglomerular feedback mechanism.31 Backleakage of fluid involves tubular factors and is not a result of hypofiltration, although it does impair the excretory function of the kidney. Consequently, a falsely low estimation of actual GFR may occur because tubular fluid containing urea and creatinine leaks back into the vascular space and interstitium. Prevention of the tubular obstruction may alter the course of renal failure, even in those states in which the primary mechanism of injury is not obstruction.
AKI may be viewed as an evolving process into three phases32: (1) an initiation phase in which the primary mechanism of injury is operational; (2) a maintenance phase during which renal function remains poor and other factors may contribute to sustained injury; and (3) a recovery phase during which there is regeneration of cells and restoration of function. Although the primary initiating event may be hypoperfusion with ischemia, often it involves multiple contributing factors that mediate additional cellular damage, usually through alterations in energy supply. From a clinical perspective, it may be most helpful to consider acute renal dysfunction syndromes according to the cause of the inciting event; however, it is equally vital to understand the other contributing mechanisms that ultimately affect outcome.
Recently, endothelial injury and vascular dysfunction have been postulated to occur during the initiation and particularly during the maintenance phases of AKI. Although most studies have focused on the tubular cell as the primary site of injury leading to dysfunction, recent studies have provided insight into the potential role for endothelial injury in continued reduced RBF and altered vascular function.33 Sutton et al. propose that an additional phase be added to the current model for AKI: after the initiation phase, an extension phase occurs that is due to microvascular injury related to ischemic damage to endothelial cells, infiltration of leukocytes, and activation of the coagulation system. This process is thought to predominate in the corticomedullary and outer medullary microvessels and may occur in the face of early tubular cell regeneration so that limiting the extension process provides a potential mechanism for aiding recovery.
Mechanisms of Renal Cell Injury
Heat shock proteins (HSPs) are a family of proteins that appear to protect cells from injury as a result of hyperthermia, ischemia, or toxins. The HSP induction by sublethal thermal stress has been found to attenuate subsequent injury in the kidney.34 Renal transplants from animals that underwent short-term hyperthermia had better initial function and subsequent survival. Furthermore, in cultured inner medullary collecting-duct cells, induction of HSP-1 by preconditioning hyperthermia attenuated the alterations in mitochondrial function and glycolysis that were observed after cells were exposed to high temperatures. Investigations into potential mechanisms to use this natural cell defense mechanism are under way.
The ability of renal tubular epithelial cells to undergo regeneration determines in large part the degree of renal recovery. Therefore much work has recently been done to study ways that cells regenerate and mechanisms that might enhance recovery. Early in ischemic injury there is induction of early response genes such as c-fos and Egr-1.35 By 2 days after ischemia, the proliferating cell nuclear antigen is detected, followed by expression of other dedifferentiated cell markers, which seem to be a sign of early recovery. Other cells appear to undergo apoptosis or cell death. Postischemic regeneration seems to be a recapitulation of early renal tubular cell development. Growth factors such as insulin-like growth factor-1 (IGF-1) and epidermal growth factor 1 (EGF-1) have been associated with enhanced recovery as well. Renal levels of hepatocyte growth factor (HGF) increase after two models of renal failure, postnephrectomy, and following CCL4 (chemokine [C-C motif] ligand 4) injection, and this increase supports a role for HGF in renal repair.36 Exogenous EGF has been shown to enhance renal tubular cell regeneration and to lessen the severity and duration of hypoxia and toxin-induced renal failure. EGF receptor levels increase within hours of ischemic injury in the rat. Elevation of soluble EGF occurs along with morphological evidence of tubular injury within 12 hours of ischemia, which is followed by cell proliferation and a decrease in soluble EGF by 24 to 48 hours after ischemia.
Alterations in Cell Membranes
Membrane phospholipids have a structural function and affect membrane permeability, as well as the activity of membrane transport systems.37 These compounds are regulated in part by the activity of phospholipases, which release free fatty acids from phospholipids. Several mechanisms related to acute cell injury may alter phospholipase activity and thereby change membrane phospholipids and membrane integrity: altered intracellular calcium homeostasis, depletion of ATP, and lipid peroxidation.33 Increased phospholipase activity has been associated with an abnormal increase in permeability of the inner mitochondrial membrane, which ultimately results in disruption of mitochondria and loss of the ability to produce adequate energy.
Cellular Calcium Homeostasis
Increased intracellular calcium is commonly found in cell injury. It is not, however, a consistent finding in all models of renal injury.38 Techniques to study changes in the subcellular distribution of calcium have allowed time-related changes to be assessed. In the rat proximal tubule, steady-state hypoxia is accompanied by a prompt increase in cytosolic free calcium that precedes the appearance of membrane damage. The increase in calcium is reversed with reoxygenation. Increased cellular calcium may activate phospholipases, as previously mentioned; alter the cytoskeleton and cause injury by allowing cell swelling; or affect membrane permeability at the plasma membrane, the mitochondrial membrane, or the endoplasmic reticulum. Alterations in mitochondrial function that occur as a result of calcium loading of this organelle have been extensively studied. Excess mitochondrial calcium is associated with changes in the permeability of the inner mitochondrial membrane with loss of the electrochemical gradient and the capacity for oxidative phosphorylation. In addition, changes in enzyme activity and mitochondrial levels of nucleotides may exist.
Production of Free Radicals
Renal cell damage induced by inflammation or oxygen deprivation may be mediated, in part, by oxygen free-radicals that are generated by several cell processes, including accumulation of long chain acyl CoA as a result of mitochondrial dysfunction. The net result is increased intracellular calcium and ultimately, changes in membrane-related functions.32
Tubular Cell Energy Metabolism
After exposure to a variety of nephrotoxins or ischemia, renal cortical ATP levels are reduced even before changes in membrane integrity and cell death occur.39 In ischemic injury, alterations in renal perfusion may result in decreased oxygen delivery to tubular epithelium. Direct mitochondrial damage has been postulated to be the primary event in many forms of nephrotoxic injury.32 Other nephrotoxins interfere with energy production by the inhibition of enzymes along the citric acid cycle. In this way, toxins impair energy production. ATP levels decrease immediately after ischemia, with concomitant increases in ATP hydrolysis products. Reflow is associated with a gradual increase in cell ATP levels.
Classification of Acute Glomerulotubular Dysfunction
Hemodynamically Mediated Acute Kidney Injury
Renal hypoperfusion with ischemia is a common form of acute renal damage, especially in the setting of the ICU. This form of renal injury is often accompanied by oliguria and results from alterations in renal perfusion after a period of hypoxia, hypotension, cardiac dysfunction, or any condition that promotes hemodynamic instability, decreased effective plasma volume, or both states. This condition is commonly referred to as acute tubular necrosis because it is characterized by necrosis of tubule cells; however, this is a nonspecific term that may also define nephrotoxic injury. A preferred term is vasomotor nephropathy or hemodynamically-mediated renal failure.40 The same physiological alterations that initiate renal injury in this form of nephropathy may potentiate renal failure in conditions whose primary inciting event may not have been vascular.
Vasomotor nephropathy commonly follows a period of renal compensatory changes that may be termed prerenal failure,9 which are discussed in the preceding section on physiology. When the kidney has fully used normal compensatory mechanisms, renal oxygen delivery is critically impaired, and this impairment results in cell damage or tubular cell necrosis. Thus it is apparent that acute tubular necrosis is the end result of a continuum of renal adaptive mechanisms. Acute cortical necrosis is an exaggerated and more advanced form of renal ischemia.
When vascular or hemodynamic abnormalities persist or are profound, renal compensatory mechanisms are unable to preserve RBF and maintain sufficient oxygen delivery and GFR. At a mean renal perfusion pressure of 80 mm Hg, afferent, arteriolar dilation is maximal, and below these systemic pressures, RBF dramatically declines.9,40 In addition, loss of the ability to autoregulate as a result of ischemia may cause further damage. Renal cell injury develops as the result of deficient oxygen delivery, depletion of cellular energy, loss of membrane integrity, and release of reactive oxygen species. Without sufficient oxygen, the kidney cannot support cell functions that maintain architectural integrity and complex transport functions.
Although total RBF is decreased in vasomotor nephropathy, outer cortical blood flow is preferentially reduced. The medulla is not spared, however, because of its increased susceptibility to alterations in renal perfusion.41 Oxygen delivery to this segment of the kidney is precarious. Medullary partial pressure of oxygen (PO2) is approximately 10 mm Hg in the rat and dog. This oxygen level approaches the critical minimum level required to support oxidative phosphorylation and ATP synthesis for cell function. In general, however, the proximal tubule sustains the greatest injury. The renal arteriogram of human subjects with vasomotor nephropathy reveals marked narrowing of the arcuate arteries and absence of peripheral vasculature, providing further evidence for the marked vascular resistance enhancement.40
The primary event in vasomotor nephropathy is injury of the renal tubule. The initiation of this injury, however, is microvascular in origin. Maximal renal compensation with marked efferent and afferent arteriolar vasoconstriction reduces glomerular plasma flow with resulting hypofiltration and compromises postglomerular blood supply to the renal tubule. Tubular cell necrosis with sloughing of tubular cells into the lumen results in obstruction of flow and backleakage of filtrate through the injured epithelium. Alterations in tubular cell function in cells receiving sublethal or lethal injury increase fluid and salt delivery distally, and this increase signals the glomerulotubular feedback system to cause vasoconstriction of the afferent arteriole and limit the fraction of plasma filtered at the glomerulus.40 Although the initial reduction in GFR is the result of decreased RBF and tubular factors such as obstruction and backleakage, continued hypofiltration during the maintenance phase is related primarily to continued vasoconstriction and renal hypoperfusion.40 Recovery from postischemic AKI is biphasic. Initially, an increase in GFR occurs with relief of tubular obstruction and subsequently, improved filtration in association with renal vasodilation.
Oliguria in the presence of renal hypoperfusion has been referred to as acute renal success by investigators who propose that the response of an intelligent organ to a perceived reduction in blood flow is to reduce fluid and electrolyte losses by vasoconstriction to reduce the fraction of plasma filtered and by maximal reabsorption of fluid and salt to restore the circulation. In addition, increased distal delivery of water and solutes because of tubular cell necrosis reflects failure of the renal tubule to absorb what is filtered. The appropriate response of an intact nephron is to reduce filtration by release of angiotensin II into the interstitium. Angiotensin II mediates arteriolar vasoconstriction, which decreases glomerular plasma flow, and retraction of the glomerular tuft, which reduces Kf, the net effect being decreased glomerular filtration.41
The classic form of hemodynamically-mediated AKI was oliguric, by definition; however, nonoliguric acute vasomotor nephropathy is increasingly recognized.41,42 This form of less severe disease has been referred to as attenuated acute tubular dsyfunction and has allowed the recognition of three stages of AKI that actually represent a continuum of worsening disease: first, abbreviated renal insufficiency occurs after a single event of renal hypoperfusion, such as aortic cross-clamping, in the face of adequate volume repletion and SBP. This syndrome is characterized by an acute drop in the GFR with gradual return to normal within a few days. The inability to concentrate the urine or to conserve sodium provides evidence of tubular injury. The second phase or form is referred to as overt renal failure. An example of this situation is aortic cross-clamping followed by continued renal hypoperfusion because of poor cardiac function. A more prolonged period of hypofiltration lasts for several days to weeks with a gradual return of the GFR. If recovery of renal perfusion is impaired by repeated episodes of ischemia/hypotension, sepsis, or hypoxia, the third pattern may be observed in which a protracted course may be observed and chances for recovery may be doubtful.42 One situation in which the last example could exist is aggressive hemodialysis (ultrafiltration) with hypovolemia and, consequently, renal hypoperfusion in the recovering phase of renal failure. Clinical experience has supported this theory. Patients with multiple renal insults have a more protracted course and increased morbidity.43
The pRIFLE classification is defined by the percent reduction in eCrCl and/or the amount of diminishing urine output2 (Table 71-1).
Table 71–1 pRIFLE Classification
| pRIFLE | Estimated CrCL | Urine Output |
|---|---|---|
| Risk | 25% decrease | < 0.5 mL/kg/h for 8 hours |
| Injury | 50% decrease | < 0.5 mL/kg/h for 16 hours |
| Failure | 75% decrease | < 0.3 mL/kg/h for 24 hours or anuric for 12 hours |
| Loss | Persistent failure >4 weeks | |
| End-stage renal disease | Persistent failure >3 months |
Treatment of Acute Kidney Injury
Prevention/Attenuation of Acute Renal Failure
Prevention or attenuation of ARF has been the subject of numerous studies, as most agree that protection of the kidney from damage or enhancing recovery after damage would be preferable to the currently available supportive therapies. Primary prevention of AKI in the ICU is limited to those conditions in which the timing of injury is predictable, such as exposure to radiocontrast dye, cardiopulmonary bypass, nephrotoxic medications, or chemotherapy. In contrast to most cases of community-acquired AKI, nearly all cases of ICU-associated AKI result from more than a single insult.46 Protective agents have been studied extensively with animal models of acute renal injury. Some of these agents have ultimately been used in clinical situations with variable success. In general, methods to reduce renal injury have been aimed at manipulation of RVR or alteration of the metabolic processes of the renal tubular cell.
Dopamine
Dopamine, when infused in low intravenous doses, increases RBF, increases GFR, and increases sodium excretion. In the past, clinicians frequently used “renal dose” dopamine in the hopes that such a maneuver might attenuate renal injury and improve survival. In addition, clinicians often interpret an increase in urinary output as proof that these two assumptions are valid. Dopamine stimulates both dopaminergic and adrenergic receptors. As such, dopamine may affect renal blood flow by direct vasodilation (dopamine receptors), by increasing cardiac output (β-receptors), or by increasing perfusion pressure. Of particular interest are its action on Dopamine 1 (D1) receptors, which are abundantly distributed throughout the renal vasculature.47 Stimulation of D1 receptors results in vasodilation by means of receptor coupling with cyclic adenosine monophosphate (cAMP) and calcium flux generated by protein kinase A. In addition, D1 receptors are also found within the brush border and basolateral membranes of the proximal tubule; medullary ascending limb of the loop of Henle; distal tubule; and cortical collecting ducts where agonist induces decreases in sodium, phosphate, and bicarbonate absorption. D1 receptors have also been localized to the macula densa where they may modify renin production.47 Dopamine inhibits the Na/K-ATPase along the nephron. Interestingly, this action would be expected to decrease the oxygen consumption of the renal tubule; thus it would be less susceptible to ischemic or hypoxic injury. Dopamine 2 (D2) receptors are present along the renal tubule. In the inner medulla, a subclass, D2k is coupled to prostaglandin E2 and attenuates the action of antidiuretic hormone (ADH) in this segment.
However, dopamine does have noncardiac effects that complicate its use and should make clinicians rethink its previous role as a vasoactive agent. The group that formulated the 2008 International Guidelines for Management of Severe Sepsis and Septic Shock does not recommend the use of low-dose dopamine as a renal agent, based on available data.48 They also noted that dopamine may inhibit thyrotropin hormone release from the hypothalamus and have immunosuppressive effects through its inhibition of release of the lymphotropic factor prolactin. Dopamine should be used cautiously in neonates because the renal vascular response to dopamine is age dependent,49 although administration of dopamine (0.5 to 2 μg/kg/min) to premature neonates with respiratory distress syndrome and renal insufficiency was reported to result in improved creatinine clearance without major side effects.
Diuretics
Intravenous diuretics have been frequently used in the intensive care unit to ameliorate fluid overload by increasing urine output.50 This widespread use of loop diuretics in the face of impending renal failure has been ascribed to a combination of animal and human data. Loop diuretics decrease renal vascular resistance and increase renal blood flow.51 In addition, loop diuretics inhibit the sodium/potassium chloride cotransporter system (NKCC2), thereby reducing active oxygen transport and potentially reducing oxygen consumption, and thus limiting ischemic injury to the outer medullary tubules. Indeed, furosemide has been shown to decrease renal oxygen consumption in critically ill patients.
Mannitol may attenuate renal failure if it is given before the insult or immediately afterward.52 Loop diuretics, such as furosemide, if given along with a potentially nephrotoxic agent, may increase the renal excretion of the agent and reduce associated nephrotoxicity. Mannitol has been shown to ameliorate nephrotoxicity related to gentamicin, amphotericin B, cisplatin, and myoglobin. A specific beneficial effect is doubtful, however, because acute saline loading alone provides similar protection. When tubular obstruction plays a major role, mannitol may increase tubular flow enough to wash obstructing debris downstream. It seems reasonable to use mannitol and potentially furosemide in the initial phases of oliguria when AKI may not be established; however, these agents provide little benefit and may increase toxicity in sustained oliguria as a result of tubular necrosis.
Calcium Entry Blockers
These agents may prevent renal insufficiency through their vasodilatory action on renal vasculature, as well as inhibition of calcium entry. The calcium channel blockers verapamil, nitrendipine, diltiazem, and nisoldipine have been administered to various animal models of ischemic injury with some success in the prevention or attenuation of renal failure. Minimal protection is observed, however, if they are administered after ischemia.52 Calcium entry blockers had a beneficial effect in endotoxin-mediated AKI. This effect was postulated to be a result of an antagonism of platelet-activating factor.53 The perfusion of cadaveric renal grafts before transplantation with diltiazem was associated with improved graft survival compared with control subjects.54 Preoperative administration of calcium channel blockers to adults undergoing cardiac surgical procedures did not provide any obvious protection from the development of AKI.55
Prostaglandins
Vasoconstrictive forces in the renal vasculature may result from the action of vasoconstrictor prostaglandins and are counteracted by the vasodilatory substances.56 Infusion or stimulation of the vasodilatory prostaglandins or inhibition of the vasoconstrictor prostaglandins seems to be a reasonable approach. Prostacyclin provided protection during ischemia in a rat model.56 Administration of the thromboxane synthetase inhibitor OKY-046 partially ameliorated hypofiltration in a rat model of ischemic renal failure.57 In addition, the administration of the free radical scavenger’s dimethylthiourea and superoxide dismutase attenuated renal insufficiency and reduced thromboxane levels.
Renin-Angiotensin Antagonists
Administration of saralasin, an angiotensin II receptor antagonist, either before or after ischemia was not beneficial in the rat model. Blockade of angiotensin production by the conversion of enzyme inhibition with enalapril or captopril was not successful in preventing AKI.58 Although captopril did prevent a fall in RBF, in one study the GFR actually dropped.
Adenosine and Adenosine Triphosphate
Renal ischemia results in the depletion of cellular adenine nucleotides and increased levels of adenosine, an agent implicated as a mediator of local renal vasoconstriction.59 Adenosine may also have protective tubular effects during ischemia because it inhibits solute reabsorption in the medullary thick ascending limb of the loop of Henle. Theophylline, a competitive inhibitor of adenosine receptors, partially prevents the hypofiltration following ischemia in the rat.
Infusion of ATP-magnesium chloride (ATP-MgCl2) after renal ischemia promotes more rapid cellular recovery and attenuates renal injury.59 In animal models exogenous ATP, adenosine diphosphate (ADP), adenosine monophosphate (AMP), and adenosine preserve renal tubular cell metabolism during anoxia by protecting the membrane from disruption and providing precursors for rapid synthesis of ATP during reperfusion.
Atrial Natriuretic Factor
Atrial natriuretic factor (ANF) has direct effects on glomerular hemodynamics and GFR.60 ANF dilates arcuate, interlobular, and proximal afferent arterioles, and it relaxes mesangial cells.61 In a rat model of rhabdomyolysis, administration of ANF improved GFR and enhanced sodium and water excretion. In addition, ANF improved GFR and maintained cell energy levels during ischemic injury.62 ANF preserves glomerular filtration and cellular ATP levels in experimental models of AKI by its effect on glomerular hemodynamics.
Thyroxine
Thyroxine reduces renal injury in a number of experimental models when given before the injury, immediately after, or 24 hours after ischemia. The mechanism by which thyroxine preserves both glomerular and tubular function is not completely understood; however, the rate of recovery of cellular ATP levels was much more rapid in animals given thyroxine after ischemic AKI. Isolated mitochondria from rats subjected to 45 minutes of ischemia exhibited decreased mitochondrial ADP transport. Administration of thyroxine was associated with significantly enhanced ADP transport.63 The investigators speculated that part of the ATP depletion associated with ischemic injury might be the result of decreased mitochondrial uptake of the ATP precursor, ADP. The administration of thyroxine at 5 to 6 ng/kg/day for 5 to 10 days in eight children with AKI resulted in the recovery of renal function in all but one child, who died of the original disease.64
Glycine and Alanine
The amino acids glycine and alanine have recently been shown to have cytoprotective effects against injury in anoxia-hypoxia and chemotherapy-induced renal failure. The mechanism of cytoprotection is not understood but does not appear to involve preservation of intracellular ATP levels. Studies performed in cultured proximal tubular cells indicate that glycine and alanine may stimulate the expression of hsp genes and increase HSP proteins, which protect cells from injury. The cytoprotective effect was not observed with other amino acids and was independent of cellular ATP levels in this model of renal injury.65 Incubation of isolated renal tubules with glycine during hypoxia was associated with increased levels of glutathione, as well as increased cell ATP, although these did not appear to account fully for the protective effect of glycine. In addition, administration of glycine prevented renal injury in rats treated with nephrotoxic doses of cisplatin.66
Acute Kidney Injury: Clinical Impact
Severe deterioration of kidney function can have a profound effect on body fluid homeostasis and on blood pressure. The nature of these alterations often requires intensive care management regardless of the precise underlying diagnosis. A wide variety of kidney diseases may result in AKI. The most urgent aspects of AKI are (1) hyperkalemia, (2) severe hypertension, (3) severe plasma and extracellular volume expansion leading to heart failure and pulmonary edema, (4) unremitting metabolic acidosis, (5) hypocalcemia/hyperphosphatemia. Each of these can be viewed as an indication for intensive care and consideration of dialysis.67 Additionally, over the past decade, the presence and degree of fluid overload (FO) has been shown to be a predictor of survival at the initiation of renal replacement therapy (RRT), and is now considered as an important indication for intervention.68,69,70,71
Plasma and Extracellular Volume Expansion
Plasma and extracellular volume expansion are associated with kidney failure. With an abrupt decline in GFR, even “normal” amounts of sodium and water intake expand the extracellular and plasma volumes. Depending on the cardiac status of the patient, the serum albumin level, and the degree of capillary permeability, this extracellular and plasma volume expansion may be manifest as peripheral edema, hypertension, or congestive heart failure and pulmonary edema. In situations of hypertension or congestive heart failure, the treatment involves two principles. The first is to reduce to as low a level as possible the amount of sodium and fluid the patient receives. This requires attention to diet, intravenous or hyperalimentation solutions, and drugs. The second principle is to remove extracellular fluid. If the patient’s kidney function permits (glomerular filtration of approximately 15 mL/min or higher), then diuretics, especially loop diuretics such as furosemide, bumetanide, or ethacrynic acid, will help to stimulate a diuresis that should improve the blood pressure or the congestive heart failure. The addition of thiazide diuretics prior to the use of loop diuretics may potentiate the effectiveness of loop diuretics allowing for a greater diuresis.72 In children with more severe kidney disease, diuretic therapy does not result in diuresis, and dialysis will be necessary.
Hypocalcemia/Hyperphosphatemia
Hypocalcemia arises from hyperphosphatemia as a result of dietary load, cellular breakdown, and reduced kidney phosphate excretion; reduced synthesis of calcitriol; downregulation of skeletal cell receptors for PTH; and acidosis.73
< div class='tao-gold-member'>
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


