122 Fluoroquinolones
 Mechanism of Action
Mechanism of Action
DNA gyrase and topoisomerase IV enzymes are thought to be essential for the replication of DNA and partition of replicated chromosomal DNA.1 DNA gyrase, a tetrameric enzyme consisting of two A and two B subunits, is known to be a primary target of fluoroquinolones in gram-negative bacteria and is the only known enzyme capable of introducing negative super-helical twists into bacterial DNA.1,2 The two subunits of gyrase are encoded by gyrA and gyrB, which are also potential sites of mutation and subsequent quinolone resistance.1,3 Topoisomerase IV seems to be a primary target of many fluoroquinolones in gram-positive bacteria such as Staphylococcus aureus and Streptococcus pneumoniae.3,4 Bacterial topoisomerase IV appears to be the principal enzyme that resolves or “decatenates” interlocked daughter DNA circles occurring at the completion of a round of DNA replication, allowing segregation of daughter chromosomes into daughter cells.1,3,4 Topoisomerase IV, like DNA gyrase, is composed of four subunits, two each of the parC and parE gene products.
As part of the topoisomerase reaction mechanism, DNA gyrase and topoisomerase IV transiently break the DNA backbone and pass a double strand of DNA through those breaks, thus introducing a negative supercoil into the DNA strand.1,2 Fluoroquinolone antibiotics have been shown to target DNA gyrase and topoisomerase IV while these enzymes are functionally attached to the DNA strand in the presence of adenosine triphosphate, resulting in a drug/enzyme/DNA complex in which the DNA remains broken.1,2 Cell death apparently results from release of double-stranded DNA breaks from multiple drug/enzyme/DNA complexes throughout the chromosome.1,2 This mechanism of action does not in itself explain why the fluoroquinolones kill bacteria so rapidly, and it has been suggested that additional protein synthesis mechanisms involving unidentified “protein factors,” interference with the “SOS” response involved in the repair of damaged DNA, dissociation of gyrase subunits, and increased oxidative stress may all play a role in the rapidly bactericidal effects of these drugs.1,2 Fluoroquinolones have also been noted to substantially decrease the synthesis of proinflammatory cytokines, although the relevance of this finding to their overall pharmacologic activity is unknown.5
 Antimicrobial Spectrum of Activity
Antimicrobial Spectrum of Activity
Fluoroquinolones have excellent in vitro activity against a wide range of both gram-positive and gram-negative organisms. Representative activities of fluoroquinolones which are currently available and frequently used in critically ill patients are shown in Table 122-1. The entire fluoroquinolone class displays excellent activity against enteric gram-negative aerobic bacteria as well as Haemophilus influenzae, Moraxella catarrhalis, and Neisseria spp. Gastrointestinal (GI) pathogens such as Salmonella spp., Shigella spp., and Campylobacter spp. are also highly susceptible to fluoroquinolones. Although some differences in relative potency exist between individual drugs as determined by the minimum inhibitory concentration (MIC) for these organisms, little difference in clinical efficacy should be expected in the treatment of infections due to susceptible strains. Activity against P. aeruginosa is more variable, however. Ciprofloxacin has traditionally been considered the most active fluoroquinolone against this organism, but data suggest there is little difference between ciprofloxacin and levofloxacin in terms of relative susceptibility of P. aeruginosa strains.6 Ciprofloxacin was active against greater than 95% of P. aeruginosa strains when first released to the market in 1987, but by 2001 both ciprofloxacin and levofloxacin were active against only approximately 65% to 80% of strains. Such high levels of resistance (25%-35%) are still seen among P. aeruginosa strains.6–10 Clinically relevant differences between ciprofloxacin and levofloxacin are further minimized when pharmacokinetic and pharmacodynamic properties are considered.9 Moxifloxacin tends to be the least active of the currently available agents.6,11–13 Nearly all fluoroquinolones adequately inhibit P. aeruginosa at concentrations achieved in the urine.
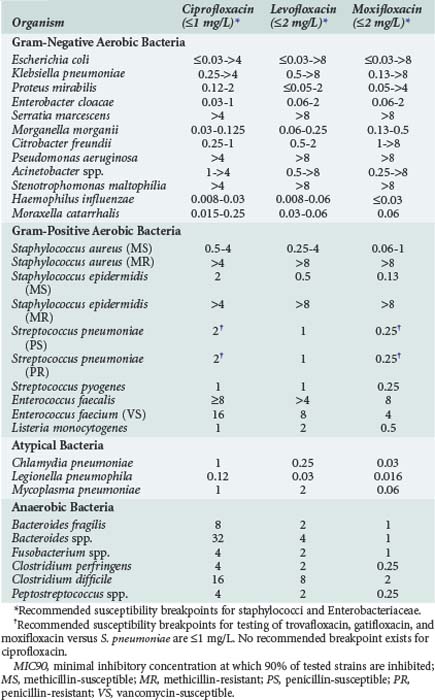
Activity of the fluoroquinolones against other hospital-acquired pathogens is also highly variable. Levofloxacin tends to be slightly more active against Acinetobacter spp., whereas moxifloxacin usually displays the best activity and ciprofloxacin is consistently the least active agent against Stenotrophomonas maltophilia.6,11–13 However, resistance to these latter organisms is quite common, and even agents with the best relative in vitro activity are not reliably clinically effective against many isolates.6,11–13
Several studies have reported that fluoroquinolones produce synergistic activity against gram-negative bacilli when used in combination with β-lactam antibiotics.14,15 These studies primarily evaluated antibiotic synergy against P. aeruginosa due to the frequent use of fluoroquinolones in antipseudomonal treatment regimens; ciprofloxacin and levofloxacin have been shown to achieve synergy against 25% to 75% of tested strains. One previous study also demonstrated synergistic in vitro activity against P. aeruginosa with the combination of moxifloxacin and either ceftazidime or cefepime15; however, additive or synergistic activity with moxifloxacin-containing combinations has not been extensively evaluated against other organisms. The ability of moxifloxacin to produce synergistic activity against P. aeruginosa may often be limited by clinically achievable drug concentrations. The use of ciprofloxacin and levofloxacin in combination regimens is most likely to result in synergistic activity, owing to their more potent activity and higher serum concentrations relative to the bacterial MICs.14,15
Newer fluoroquinolones have improved activity against gram-positive bacteria relative to older agents such as ciprofloxacin. Moxifloxacin has the best overall activity against staphylococci and streptococci, followed by levofloxacin and more distantly by ciprofloxacin.12,13 Levofloxacin and moxifloxacin are reliably active against penicillin-susceptible strains of S. pneumoniae; this activity is also retained against strains of S. pneumoniae resistant to other drug classes including penicillins, macrolides, and sulfonamides. Although ciprofloxacin has only moderate activity against methicillin-susceptible S. aureus (MSSA), newer agents have excellent activity against this organism. None of the fluoroquinolones is reliably active against methicillin-resistant S. aureus (MRSA), and rates of fluoroquinolone resistance among MRSA are quite high. Fluoroquinolones as a class also have only moderate activity against enterococci, with great variability seen among the various agents and specific bacterial strains.11–13 Fluoroquinolones have consistently excellent activity against Listeria monocytogenes.11–13
The activity of various fluoroquinolones against anaerobic bacteria is highly variable. Trovafloxacin was the first commercially available fluoroquinolone with clinically relevant anaerobic activity in vitro, as well as proven clinical efficacy for anaerobic infections including complicated intraabdominal infection. Moxifloxacin has in vitro activity against Bacteroides fragilis, Bacteroides group organisms, Fusobacterium spp., Clostridium spp., and other anaerobes that is generally comparable to trovafloxacin.12,13,16 However, recent data have demonstrated resistance rates in excess of 30% for many clinically important anaerobes, and the appropriateness of moxifloxacin for treatment of serious anaerobic infections is questionable.16 Neither ciprofloxacin nor levofloxacin have clinically relevant activity against anaerobic bacteria.
 Mechanisms of Fluoroquinolone Resistance
Mechanisms of Fluoroquinolone Resistance
Two basic mechanisms of fluoroquinolone resistance have been identified. One involves alteration of DNA gyrase and topoisomerase IV, whereas the other results in reduced drug accumulation within bacterial cells.2,17 Plasmid-mediated resistance, once considered quite rare, appears to be spreading rapidly among enteric gram-negative bacilli in certain geographic regions.18 However, plasmid-mediated resistance is relatively unusual compared with the more typical chromosomally mediated mechanisms of resistance.
Mutations in quinolone-resistance determining regions (QRDR) of topoisomerase enzymes prevent formation of drug/enzyme/DNA complexes, allowing DNA synthesis to occur in the presence of the drugs. Mutations in the genes encoding DNA gyrase (gyrA and gyrB) have been most frequently identified. However, other quinolone-resistant mutations in parC and parE, the genes encoding topoisomerase IV, have also been identified.1,2,17,19 Resistance to fluoroquinolones appears to arise in a stepwise manner. In some species (e.g., gram-negative bacteria), first-step mutations occur in gyrA and occasionally in gyrB, whereas in other species (e.g., S. aureus, S. pneumoniae) first-step mutations occur in parC and less often in parE.17,19 First-step mutations usually result in a low-level resistance (≤fourfold increased MIC), whereas additional mutations in either primary or secondary enzyme targets (second-step mutations) result in high-level resistance to drugs at clinically relevant concentrations. Dual gyrA and parC mutations have been described in clinical isolates of S. pneumoniae; however, it is thought that these strains were selected by fluoroquinolones with less potent antipneumococcal activity (e.g., ciprofloxacin).19–22
The first efflux system for quinolones was identified in Escherichia coli,23 whereas the first evidence for actual efflux-mediated quinolone resistance came from the characterization of S. aureus with overexpression of the norA gene product, a protein that mediates efflux.24 Such efflux may occur in both quinolone-resistant and quinolone-susceptible strains of S. aureus. In some species (e.g., P. aeruginosa), at least two different efflux systems may be present that mediate resistance to multiple other drug classes in addition to fluoroquinolones.25 Although most efflux proteins appear to be relatively nonspecific multidrug transporters whose substrates include hydrophilic fluoroquinolones as well as monocationic organic compounds, relatively substrate-specific efflux pumps have also been described.26
Many other genetic mutations have been described that result in decreased intracellular accumulation of fluoroquinolones and low-level drug resistance. Nearly all these mutations are associated with decreased expression of OmpF, a nonspecific outer membrane porin channel that is a major route of passage of hydrophilic fluoroquinolones through bacterial cellular membranes into the periplasmic space.27 Although decreased membrane permeability is relatively common and easily induced, this is an unusual mechanism for clinically significant resistance. Strains of S. pneumoniae, E. coli, S. aureus, and P. aeruginosa have been identified that possess both altered outer membrane permeability and gyrA mutations, resulting in high-level resistance to all tested fluoroquinolones.28,29 Strains of highly ciprofloxacin-resistant Salmonella with both outer membrane protein alterations and expression of efflux pumps have also been described.30
Fluoroquinolone resistance among pathogens such as S. aureus and P. aeruginosa has been particularly problematic since the introduction of these agents into clinical use. As fluoroquinolones have become more extensively used in the treatment of respiratory tract infections, reports of increasing resistance among S. pneumoniae have focused attention on newly recognized mechanisms of drug action and drug resistance.19–22 Resistance to fluoroquinolones has tended to emerge rapidly in bacteria with lower intrinsic susceptibility (e.g., S. aureus, P. aeruginosa, and Acinetobacter spp.) because potentially fewer mutational steps are required to confer clinically relevant MIC changes.26,31,32 However, fluoroquinolone resistance has also been noted to be an increasing problem among gram-negative bacilli such as Enterobacter spp., Klebsiella pneumoniae, and even E. coli, organisms that were originally considered to be highly susceptible to the drugs.6,8,10 This problem is particularly an issue among isolates from ICUs.10 Development of resistance is also accelerated by the use of drugs with lower in vitro activity, use of inappropriately low doses to treat infections caused by less susceptible organisms, and treatment of infection at sites where quinolone penetration may be decreased.31–34 Development of resistance to one fluoroquinolone usually causes decreased susceptibility to all other agents in the class, although clinically relevant resistance may not necessarily occur. Of note, fluoroquinolone use has also been associated with high rates of cross-resistance among drugs of unrelated antibiotic classes such as the carbapenems, cephalosporins, and aminoglycosides.33–35 Although not well understood, such cross-resistance is probably mediated by up-regulation and/or reduction in multiple efflux pump systems involved in passage of antibiotics through cell membranes and intracellular drug accumulation.33–35 Although the fluoroquinolones remain highly active and clinically effective against a wide variety of important pathogens found in critically ill patients, increasing resistance is clearly an important issue in the clinical use of these drugs.
 Pharmacokinetics
Pharmacokinetics
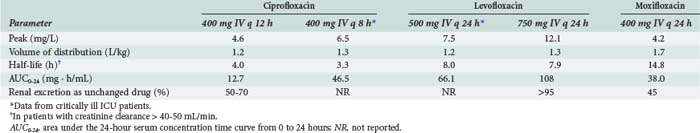
Pharmacokinetic properties of the currently used fluoroquinolones are shown in Table 122-2. Individual agents in the class exhibit distinct differences in properties such as oral bioavailability, half-lives, extent of metabolism, and routes of excretion. However, fluoroquinolones as a whole are characterized by rapid oral absorption and extensive distribution into many fluids and tissues, resulting in concentrations that are well above the MIC for many gram-negative and gram-positive organisms; serum half-lives are sufficiently long to allow once- or twice-daily dosing. Ciprofloxacin and levofloxacin have been most extensively studied in critically ill patients. Although large interpatient variability and some differences in mean parameters were observed compared with normal volunteers, pharmacokinetics of the drugs were generally similar enough to allow the use of normally recommended doses.36,37
< div class='tao-gold-member'>
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


