Fig. 12.1
The molecular structure of etomidate. Etomidate has a single chiral carbon. The R enantiomer (shown) is the more potent form and the one used clinically
The initial characterization of etomidate by Janssen Pharmaceuticals utilized a racemic mixture of the drug. Upon synthesis and study of its two enantiomers, it became evident that essentially all of etomidate’s hypnotic activity is mediated by the R enantiomer and that the therapeutic index of that enantiomer is approximately twice as high as that of the racemic mixture [4]. Consequently, it was etomidate’s R enantiomer that was ultimately developed for clinical use.
Etomidate’s comparatively high therapeutic index (defined in unventilated animals) is almost certainly attributable to its relatively modest effects on respiratory function. Studies of etomidate in humans show that it produces little or no change in respiratory parameters when given alone at clinically relevant doses [5, 6]. When given along with remifentanil, etomidate produces less respiratory depression than propofol [7]. Studies utilizing steady-state end-tidal carbon dioxide concentrations revealed that although an induction dose of etomidate (0.3 mg/kg) decreases the ventilatory drive in response to changing levels of carbon dioxide (i.e., reduces the slope of the carbon dioxide concentration—ventilatory response curve), it doubles minute ventilation when end-tidal carbon dioxide is held constant at 46 mmHg [8]. For these reasons, etomidate may be preferable to other hypnotic agents when spontaneous ventilation is desired [9–11].
Cardiovascular function is similarly preserved after etomidate administration. Etomidate does not release histamine and maintains hemodynamic stability even in patients with valvular and coronary artery disease [12–14]. As a consequence, its greatest value may be as an anesthetic induction agent for patients with significant cardiac disease. Etomidate also maintains cerebral perfusion pressure while reducing cerebral metabolic requirements and blood flow, features that are desirable in patients with space-occupying intracranial lesions [15]. In dogs, hepatic arterial and portal blood flow is reduced by etomidate [16]. Nevertheless in humans, etomidate minimally affects the metabolism of coadministered drugs [17].
It has been reported that etomidate can enhance focal epileptogenic activity in patients with epilepsy and produce generalized epileptiform electroencephalographic activity in patients without a history of seizure disorders [18]. As a sedative-hypnotic agent for electroconvulsive therapy , etomidate induces seizures that are longer in duration and with fewer side effects than methohexital [19]. For this reason, etomidate has often been advocated for patients undergoing electroconvulsive therapy and, in particular, those who are seizure resistant [20–22]. Bispectral (BIS) index values of patients who have received etomidate correlate with predicted blood concentrations. However, with etomidate, loss of consciousness tends to occur at BIS values that are lower than those seen with other anesthetic agents including propofol and sevoflurane [23].
Etomidate Pharmacokinetics and Pharmacodynamics
Anesthetic induction upon administering a typical bolus dose of etomidate (0.3 mg/kg) occurs within seconds and lasts 3–5 min [24]. With such bolus dosing, plasma etomidate concentrations reach a peak value of approximately 300 ng/ml before falling in a triexponential manner. Analysis using a three-compartment open pharmacokinetic model yields an initial half-life after bolus administration of 2.6 ± 1.3 min, an intermediate half-life of 28.7 ± 14 min, and an apparent elimination half-life of 4.6 ± 2.6 h. The two early exponential phases are consistent with etomidate’s rapid distribution into the brain with resultant anesthetic induction followed by its extensive redistribution into other tissues leading to anesthetic emergence. The late exponential phase, which defines etomidate’s elimination half-life, reflects the slow, rate-limiting return of the drug from the deep peripheral compartment and hepatic elimination. Although compartmental models do not define each compartment in terms of an actual biological organ or tissue, it seems likely that fat contributes significantly to that deep compartment [4]. Etomidate is metabolized in the liver by hepatic esterases to a carboxylic acid metabolite that has no known pharmacological activity and is excreted primarily in the urine [25].
With prolonged continuous etomidate infusion, hypnotic recovery is significantly slower than after single bolus administration. When infused for 30 ± 26 min as the hypnotic component of a total intravenous anesthetic regimen for gynecological surgery, emergence occurred (on average) 20 ± 9 min after terminating the etomidate infusion, and patients remained drowsy for 4–6 h after leaving the recovery room [26]. Hebron et al. reported that awakening from even moderate levels of sedation and steady-state plasma concentrations averaging only 158 ± 36 ng/ml occurred approximately 40 min after discontinuing etomidate infusions lasting 2 days [27, 28]. As expected from its several hour terminal elimination half-life, etomidate concentrations in the plasma remained measurable (as high as 150 ng/ml in one patient) for 24 h after terminating such prolonged infusions.
Studies in humans and animals have sought to define how pathological states modify etomidate pharmacokinetics. Human studies have shown that etomidate’s clearance rate is unaffected by hepatic cirrhosis . However, etomidate’s terminal elimination half-life is prolonged (to 9 h) because its volume of distribution is increased [29]. Elderly patients or those with hepatic or renal failure may require lower dosing because plasma protein binding is reduced [30, 31]. Studies in pigs have shown that hemorrhagic shock only minimally affects etomidate pharmacokinetics. The only notable change was a reduction in the volume of the two peripheral compartments, which likely reflected redistribution of blood flow away from these compartments as cardiac output was reduced.
Suppression of Adrenocortical by Etomidate
The most notable and potentially serious side effect of etomidate administration is the suppression of adrenocortical steroid synthesis. The ability of etomidate to suppress stress-induced steroid synthesis was first reported in 1982 by Preziosi and Vacca, but its clinical significance at the time was unclear [32]. However in their seminal letter to Lancet the following year, Ledingham and Watt reported that critically ill multiple trauma patients in their intensive care unit who received prolonged etomidate infusions for sedation had a mortality that was almost threefold higher than those who received benzodiazepines (69 % versus 25 %, p < 0.0005) [33]. They postulated that this increased death rate was attributable to etomidate’s suppression of adrenocortical function. In vitro studies using adrenocortical cells and in vivo studies in both surgical and nonsurgical patients showed that etomidate inhibited the adrenocortical cytochrome P450 enzyme 11α-hydroxylase and blocked the conversion of 11-deoxycortisol to cortisol [34–37]. After a single bolus dose of etomidate, this adrenocortical suppression lasts approximately 6 h in healthy individuals [38, 39]. However in the critically ill, such suppression can last for days [40–42].
Prior to the publication of the Ledingham and Watt letter and the subsequent full report of the study, etomidate was enthusiastically recommended as a continuously infusible sedative because it maintained respiratory drive and hemodynamic stability [33, 43–46]. Since that time, the use of prolonged etomidate infusions has been almost completely abandoned out of concerns that it increases morbidity and mortality. An interesting exception is in patients with Cushing’s syndrome where low-dose etomidate infusions continue to be used to reduce cortisol synthesis and ameliorate the symptoms of hypercortisolemia [47–51]
Etomidate continues to be used as a single bolus to induce anesthesia at the start of surgery, particularly in the elderly and critically ill. However, even this limited use is controversial as it is known to suppress adrenocortical function for many hours or even days [52–56]. Clinical studies and meta-analyses aiming to evaluate the impact of a single dose of etomidate on morbidity and mortality in the critically ill have yielded apparently conflicting results with some suggesting significant deleterious effects [57–62] and others suggesting no affect at all [63–66]. Thus, we are in a state of clinical equipoise with respect to the use of single-dose etomidate in the critically ill .
Other Adverse Effects of Etomidate
Involuntary movements (myoclonus) are commonly observed after etomidate administration, with some studies reporting an incidence as high as 80 % in unpremedicated patients [67–70]. Such movements may resemble myoclonic seizures, but are physiologically distinct. Although the mechanism of etomidate-induced myoclonus is not clear, it has been suggested that it occurs because etomidate depresses inhibitory neural circuits in the central nervous system sooner and at lower concentrations than excitatory circuits [70]. Regardless of the mechanism, myoclonus can be significantly reduced or completely prevented by administering a variety of drugs with central nervous system depressant effects including opiates [71, 72], benzodiazepines [73], dexmedetomidine [74], thiopental [74], lidocaine [75], and magnesium [76].
Pain at the injection site is another common side effect of etomidate administration, although its incidence is highly dependent upon the size of the vein into which it is injected [77] and the formulation that is used. When etomidate is formulated in either cyclodextrin [78] or a lipid emulsion of medium- and long-chain triglycerides [79, 80], the incidence of injection pain is significantly less than when dissolved in a 35 % propylene glycol/water mixture. The underlying mechanism for such pain may be etomidate’s ability to activate transient receptor potential (TRP) ion channels present in sensory neurons [81]. By lowering the free-aqueous etomidate concentration and/or reducing solution osmolality, lipid emulsion and cyclodextrin formulations may reduce TRP channel activation, leading to less pain on injection.
Finally, postoperative nausea and vomiting have long been associated with etomidate administration with reported incidences as high as 40 % [77, 82, 83]. However, a prospective study using etomidate formulated in a lipid emulsion found an incidence of nausea that was no greater than that produced by propofol perhaps suggesting that the emetogenic trigger in etomidate is the propylene glycol solvent and not the anesthetic itself. [84]
Molecular Mechanisms of Etomidate Action
Etomidate produces sedation and hypnosis by binding to and enhancing the function of GABAA receptors . This conclusion is strongly supported by numerous studies including those demonstrating high correlations between the in vivo hypnotic potencies of etomidate and etomidate analogues and their in vitro GABAA receptor enhancing activities [85–87]. They are further supported by studies showing that an amino acid mutation that reduces etomidate’s ability to enhance GABAA receptor function also reduces the sensitivity of transgenic mice containing that mutation to etomidate’s hypnotic and immobilizing actions [88]. In electrophysiological experiments, GABAA receptor enhancement may be detected at low etomidate concentrations as an increase in the receptor’s sensitivity to GABA (i.e., a leftward shift in the GABA concentration-response curve), a phenomenon commonly termed agonist potentiation. At high concentrations, etomidate can also directly activate GABAA receptors even in the absence of GABA. Site-directed mutagenesis and modeling studies by Forman et al. suggest that both agonist potentiation and direct activation result from etomidate binding to the same class of sites on the GABAA receptor’s open channel state [89, 90].
The etomidate-binding sites on GABAA receptors responsible for enhancement are thought to be located within the receptor’s transmembrane domain at the interfaces between the α- and β-subunits. GABAA receptors containing α2– or α3-subunits are more sensitive to etomidate’s enhancing actions than those containing the α1-subunit [86, 91]. This subunit selectivity may be contrasted with that exhibited by propofol, which modulates GABAA receptors containing α1-, α2-, or α3-subunits with equal potency [86]. It is, therefore, tempting to speculate that at least some of the pharmacodynamic differences between etomidate and propofol (e.g., incidence of myoclonus) are due to their differing selectivities for the various GABAA receptor subtypes present in the central nervous system. In addition to the enhancing sites, there is also a distinct site(s) that causes channel inhibition. This inhibitory site lacks the enantiomeric selectivity exhibited by the enhancing site and almost certainly binds etomidate with much lower affinity [86]. Therefore, it is unlikely that this site contributes to etomidate’s pharmacology when the drug is given at clinically relevant doses.
Rapidly Metabolized Etomidate Analogues
The prolonged adrenocortical suppression produced by etomidate results from two important aspects of etomidate’s pharmacology. First, etomidate is approximately 100-fold more potent a suppressor of adrenocortical function than it is a sedative-hypnotic [24, 92, 93]. Consequently, an anesthetic induction dose of etomidate represents a massive overdose with respect to its ability to suppress adrenocortical function. Second, etomidate’s terminal elimination half-life is rather long: 3–5 h [24, 29]. Thus, after just a single anesthetic induction dose of etomidate, many hours must pass before etomidate’s concentration in the blood falls below that which suppresses adrenocortical steroid synthesis. It is within this mechanistic context that the strategy emerged to design rapidly metabolized analogues of etomidate [94]. The premise was that because these “soft” analogues would be metabolized much more rapidly than the “hard” drug (i.e., etomidate) upon which they are based, recovery from adrenocortical suppression would occur significantly more quickly (Fig. 12.2). In addition, it was hypothesized that hypnotic recovery would similarly occur more quickly—particularly after prolonged infusion—because of this more rapid elimination.
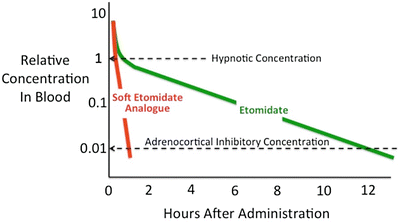
Fig. 12.2
Time-dependent change in hypnotic drug concentrations after single intravenous bolus administration. After administering a hypnotic dose of etomidate, the concentration of etomidate in the blood must fall by two orders of magnitude before adrenocortical function recovers. Because etomidate is relatively slowly metabolized, such recovery requires many hours (12 h in this figure). Soft etomidate analogues are designed to be rapidly metabolized to accelerate both hypnotic and adrenocortical recoveries
Methoxycarbonyl Etomidate (MOC-Etomidate ): The Prototypical Soft Etomidate Analogue
MOC-etomidate was the first soft etomidate analogue to be synthesized and studied (Fig. 12.3a) [94]. Similar to remifentanil and esmolol, it contains a metabolically labile ester moiety that is attached to the pharmacophore via a two-carbon spacer. Thus, it was anticipated that MOC-etomidate would be rapidly hydrolyzed by nonspecific esterases to a carboxylic acid metabolite that possessed significantly less pharmacological activity than the parent compound, MOC-etomidate.
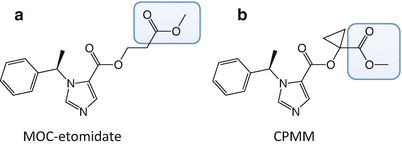
Fig. 12.3
The molecular structures of (a) methoxycarbonyl etomidate (MOC-etomidate) and (b) cyclopropyl-methoxycarbonyl metomidate (CPMM). The metabolically labile ester moiety in each etomidate analogue is highlighted by the blue box
Initial studies of MOC-etomidate in both tadpoles and rats demonstrated that similar to etomidate, MOC-etomidate rapidly induces hypnosis (albeit with potency that is 1/5 to 1/10 that of etomidate), has a high therapeutic index, and enhances GABAA receptor function. However, compared to etomidate, it is much more rapidly metabolized. For example, in pooled human liver s9 fraction, MOC-etomidate was hydrolyzed with a half-life of 4.4 min, whereas etomidate was not measurably hydrolyzed even after 40 min. Therefore, after single bolus administration, MOC-etomidate is ultrashort acting as a hypnotic and does not produce prolonged adrenocortical suppression.
In the soft analogue strategy of drug design, the pharmacological activity of the metabolite is a critical determinant of drug pharmacology. Ideally, the metabolite would have no activity at all, but in practice this is rarely the case. In the case of MOC-etomidate , the carboxylic acid metabolite that forms upon hydrolysis has potencies for activating GABAA receptors, producing hypnosis, and inhibiting cortisol synthesis that are ~300-fold less than those of MOC-etomidate [95]. Although this potency difference between metabolite and parent drug is sufficiently large to achieve the goal of rapid recovery after bolus administration and similar to that of esmolol (also 300-fold), it is less than those reported for both remifentanil (4600-fold) and remimazolam (1600-fold) [96–98].
Subsequent studies focused on assessing the potential value of MOC-etomidate as a hypnotic maintenance agent [99]. MOC-etomidate was infused into rats for periods of time ranging from 5 to 30 min. After the infusion ended, the time required for rats to recover was measured using electroencephalographic (burst suppression ratio) and behavioral (loss of righting reflexes) endpoints. After 5-minute infusions, the electroencephalogram recovered to near baseline values and righting reflexes returned in 1–2 min. However, with longer MOC-etomidate infusion times, recovery times were dramatically longer. Such marked context sensitivity was unexpected because it had not been observed with other commonly used soft drugs. Chemical analysis of the blood and cerebrospinal fluid revealed that during MOC-etomidate infusions, MOC-etomidate metabolite concentrations reached levels sufficient to cause hypnosis. After MOC-etomidate infusions ended, metabolite concentrations decreased on the timescale of hours, paralleling the time required for electroencephalographic and behavioral recoveries. This strongly suggested that the delayed recovery after prolonged MOC-etomidate infusions was caused by a large accumulation of the weakly active metabolite in the central nervous system. Thus, MOC-etomidate provided a proof of principle that soft etomidate analogues could be produced. However, its relatively low potency and very rapid metabolism required the administration of extremely large doses to maintain hypnosis and resulted in sufficient metabolite accumulation to markedly delay recovery. This made the drug unsuitable for clinical development as a continuously infused hypnotic maintenance agent.
Cyclopropyl-Methoxycarbonyl Metomidate (CPMM ): A Second-Generation Soft Etomidate Analogue
To ameliorate the problem of metabolite accumulation during prolonged infusion, work was begun to develop second-generation soft etomidate analogues with optimized pharmacokinetic and pharmacodynamic properties [100]. These efforts focused on modifying the structure of the spacer that links the metabolically labile ester moiety to the etomidate pharmacophore. It was hypothesized that the hydrolysis rate could be beneficially slowed (thus reducing infusion requirements and metabolite accumulation) by adding substituent groups onto that spacer to modestly increase steric hindrance. At the same time, it was hoped that such structural modifications would increase hypnotic potency. Thirteen new soft etomidate analogues were produced whose spacers were either one or two carbons in length and contained various aliphatic-protecting groups to slow hydrolysis. In vitro and in vivo studies revealed that the metabolic half-lives in rat blood and hypnotic potencies in rats ranged by more than an order of magnitude. The most promising of these compounds was cyclopropyl-methoxycarbonyl metomidate (CPMM). It differs from MOC-etomidate in that its spacer is only one carbon in length and contains a cyclopropyl substituent group (Fig. 12.3b). Compared to MOC-etomidate, its hypnotic potency in rats is eightfold higher, and it is metabolized by esterases slightly more slowly. Because of these pharmacodynamic and pharmacokinetic differences, infusion dosing of CPMM is one to two orders of magnitude lower than that of MOC-etomidate.
Studies in both rats and dogs showed that hypnotic recovery after terminating a continuous infusion of CPMM occurs within several minutes regardless of the infusion duration [101, 102]. Such context-insensitive recovery is distinct from that exhibited by either etomidate or propofol. Adrenocortical function similarly recovers rapidly after infusion termination. In rats, adrenocorticotropic hormone-stimulated plasma corticosterone concentrations returned to baseline values within 30 min after terminating a 2-h CPMM infusion. In dogs, adrenocortical responsiveness 90 min after terminating a 2-h CPMM infusion was equivalent to that observed after terminating a 2-h propofol infusion, suggesting that with CPMM infusions lasting only a few hours, any adrenocortical suppression was likely to be clinically unimportant.
Pharmacokinetic studies in dogs confirmed that CPMM is rapidly hydrolyzed to the expected carboxylic acid metabolite in vivo [102]. After a single CPMM bolus dose, venous plasma concentrations rapidly increased and then fell by nearly three orders of magnitude over the subsequent hour. CPMM’s terminal elimination half-life was 16.1 ± 2.99 min, which was significantly faster than that of etomidate (60.1 ± 10.4 min). The clearance of CPMM was not only significantly faster than that of etomidate (211 ± 26 versus 14.7 ± 2.78 ml · kg−1 min−1, respectively); it was also more than an order of magnitude greater than the total hepatic blood flow. This implied that the primary site of CPMM metabolism in these dogs was extrahepatic. After 2-h infusions, CPMM’s terminal elimination half-life was sixfold longer (96.8 ± 38.5 min) than that observed after single bolus administration and essentially identical to that of etomidate (88.1 ± 14.1 min). The similar terminal elimination half-lives of CPMM and etomidate after 2-h infusions suggest a common rate-controlling step. Previous pharmacokinetic modeling studies of etomidate indicate that this step is the slow return of drug into the central compartment from a poorly perfused deep compartment (e.g., fat) into which the drug had accumulated during the infusion period [24].
So how does CPMM compare with propofol? Both anesthetics enhance the function of GABAA receptors [103]. However, CPMM retains etomidate’s ß-subunit selectivity . This suggests that CPMM and etomidate act on the same GABAA receptor subtypes. In tadpoles, an animal common model system for quantifying the hypnotic potencies of anesthetic agents, CPMM was found to be approximately half as potent as propofol with median hypnotic concentrations of 2.6 ± 0.19 μM and 1.3 ± 0.04 μM, respectively [103]. However, CPMM’s median hypnotic dose in rats is approximately 1/6 that of propofol. This apparent discrepancy likely relates to the different extents to which CPMM and propofol bind to plasma proteins; protein binding is expected to be ~75 % for CPMM (i.e., similar to etomidate) compared to 98 % for propofol. Encephalographic recovery times in rats following propofol infusion are highly dependent upon the infusion duration (Fig. 12.4). For example, the time required for the burst suppression ratio to recover to within 10 % of baseline was 9.7 ± 2.3 min after a 5-min propofol infusion but 45 ± 11 min after a 120-min infusion. In contrast and as noted above, recovery after CPMM infusion is short and independent of infusion duration (Fig. 12.5). Such infusion duration-independent recovery is similar to that seen with remifentanil, which is also rapidly metabolized by nonspecific esterases to an essentially inactive carboxylic metabolite.
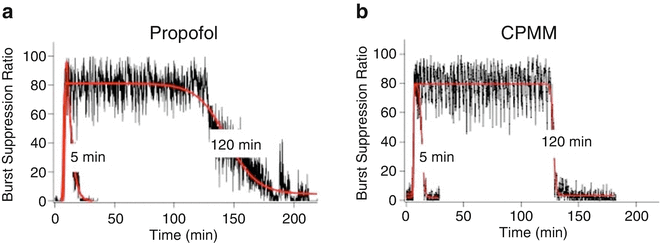
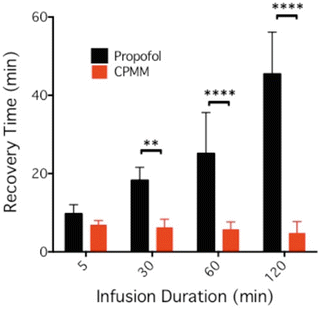
Fig. 12.4
Change in the electroencephalographic burst suppression ratio in individual rats upon infusion of (a) propofol or (b) cyclopropyl-methoxycarbonyl metomidate (CPMM). Each hypnotic agent was administered using a closed-loop system to maintain the burst suppression ratio at 80 % for either 5 or 120 min (in the presence of 1 % isoflurane). The red curves are fits of each data set to a biphasic sigmoidal equation
Fig. 12.5
90 % electroencephalographic burst suppression ratio recovery time in the rat as a function of propofol or CPMM infusion duration. This recovery time is defined as the time when the infusion ended until the time that the burst suppression returned to within 10 % of the post-infusion baseline value. The data shows that recovery times increased with infusion duration with propofol but not with CPMM. For infusion times of 30 min and longer, recovery times were significantly shorter after infusion of CPMM versus propofol. Each bar is the mean (±SD) obtained using five rats. Studies were performed in a background of 1 % isoflurane. ****p < 0.0001; **p < 0.001
Studies in rats suggest that CPMM may offer important advantages over etomidate in the setting of sepsis. In a lipopolysaccharide inflammatory model of sepsis, plasma corticosterone concentrations in rats were up to ninefold higher during 1-h infusions of CPMM as compared to etomidate suggesting that CPMM’s intrinsic ability to inhibit 11β-hydroxylase is significantly lower than that of etomidate [104]. In addition, plasma cytokine concentrations were lower, metabolic derangement was less, and survival was higher in rats that received CPMM as compared to those that received etomidate.
Under the name ABP-700, CPMM has recently completed phase 1 clinical studies in healthy human volunteers. Although the results have not been formally reported, public comments made by The Medicines Company indicate that ABP-700’s pharmacology in humans mirrors that seen in animals with an onset and offset of hypnotic action that are fast and effects on adrenocortical steroid synthesis that are similar to that seen with propofol.
Pyrrole Etomidate Analogues
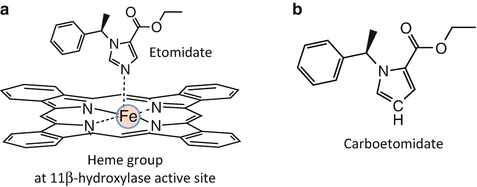
X-ray crystallographic studies of binding of imidazole-containing molecules to cytochrome P450 enzymes indicate that high-affinity binding requires a coordination bond between the basic nitrogen in the imidazole ring and the heme iron located at the enzyme’s active site [105–107]. Because 11β-hydroxylase is a cytochrome P450 enzyme, it was hypothesized that etomidate’s high binding affinity to this enzyme similarly resulted from analogous coordination bonding (Fig. 12.6a). Thus, it was predicted that binding affinity could be substantially reduced (and adrenocortical suppression eliminated) by replacing this nitrogen with other atoms that are incapable of forming coordination bonds. At the same time, it was hoped that such a molecular change would not greatly impact GABAA receptor potency (and thus hypnotic activity) and preserve etomidate’s beneficial properties such as its minimal affects on cardiovascular and respiratory function and its high therapeutic index.